

Critical Assessment of MetaProteome Investigation (CAMPI): a multi-laboratory comparison of established workflows
- PMID: 34911965
- PMCID: PMC8674281
- DOI: 10.1038/s41467-021-27542-8
Critical Assessment of MetaProteome Investigation (CAMPI): a multi-laboratory comparison of established workflows
Abstract
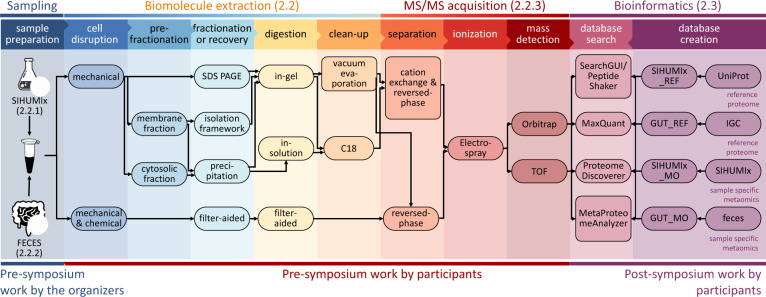
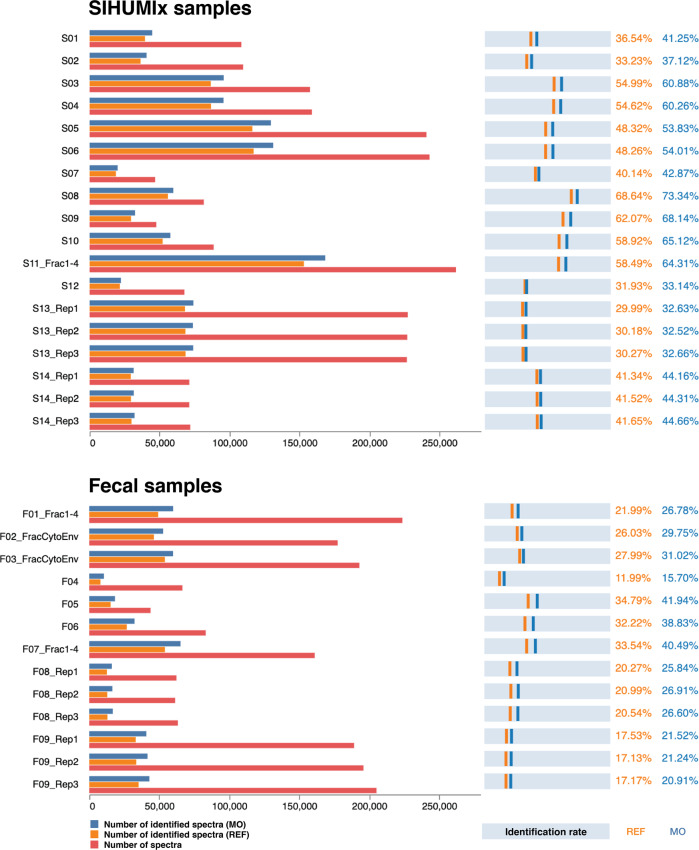
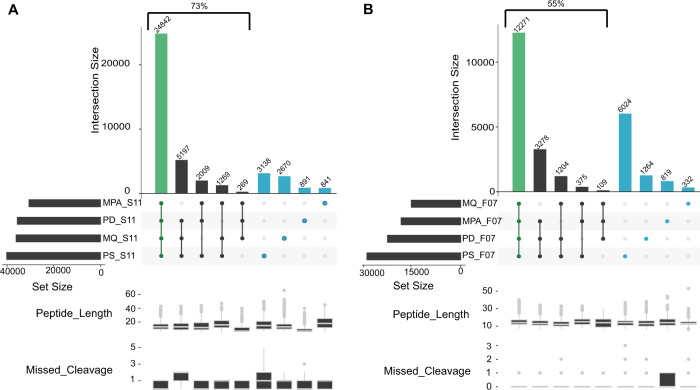
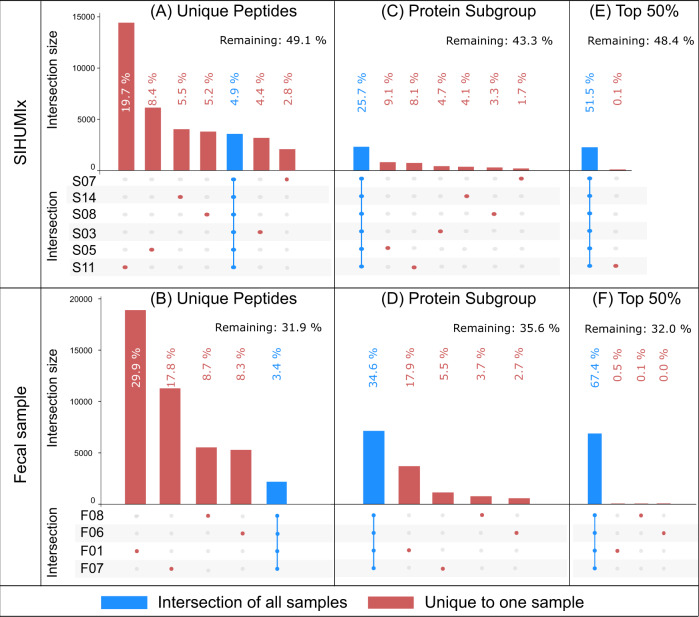
Metaproteomics has matured into a powerful tool to assess functional interactions in microbial communities. While many metaproteomic workflows are available, the impact of method choice on results remains unclear. Here, we carry out a community-driven, multi-laboratory comparison in metaproteomics: the critical assessment of metaproteome investigation study (CAMPI). Based on well-established workflows, we evaluate the effect of sample preparation, mass spectrometry, and bioinformatic analysis using two samples: a simplified, laboratory-assembled human intestinal model and a human fecal sample. We observe that variability at the peptide level is predominantly due to sample processing workflows, with a smaller contribution of bioinformatic pipelines. These peptide-level differences largely disappear at the protein group level. While differences are observed for predicted community composition, similar functional profiles are obtained across workflows. CAMPI demonstrates the robustness of present-day metaproteomics research, serves as a template for multi-laboratory studies in metaproteomics, and provides publicly available data sets for benchmarking future developments.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
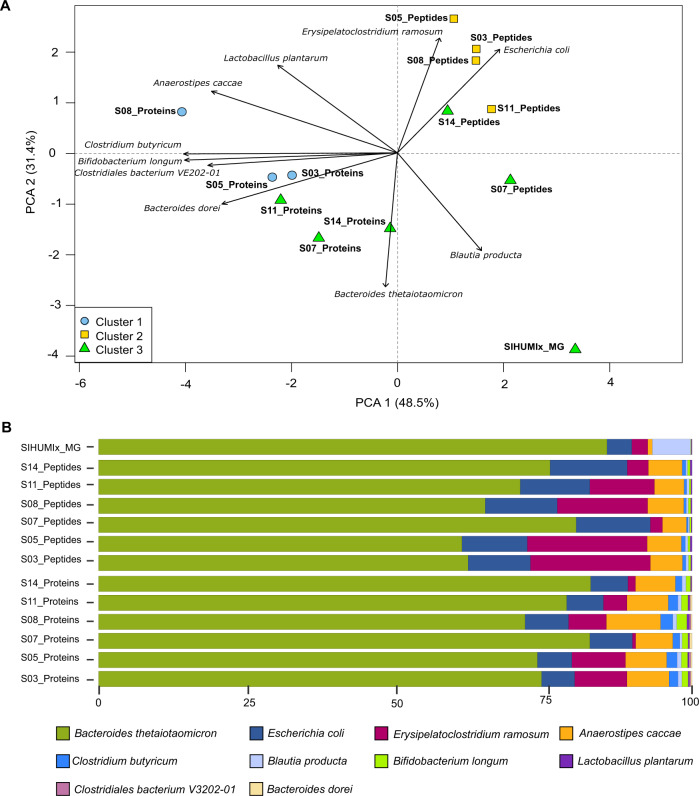
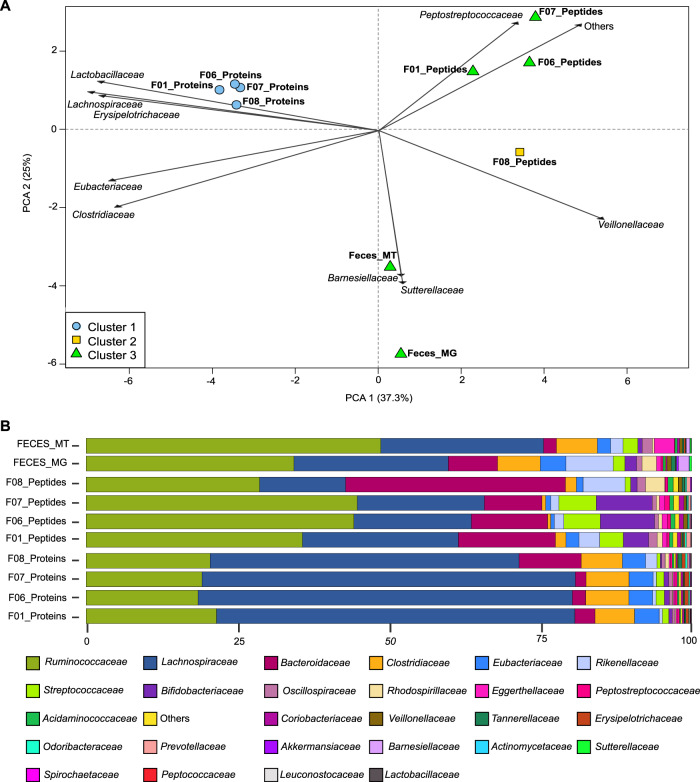
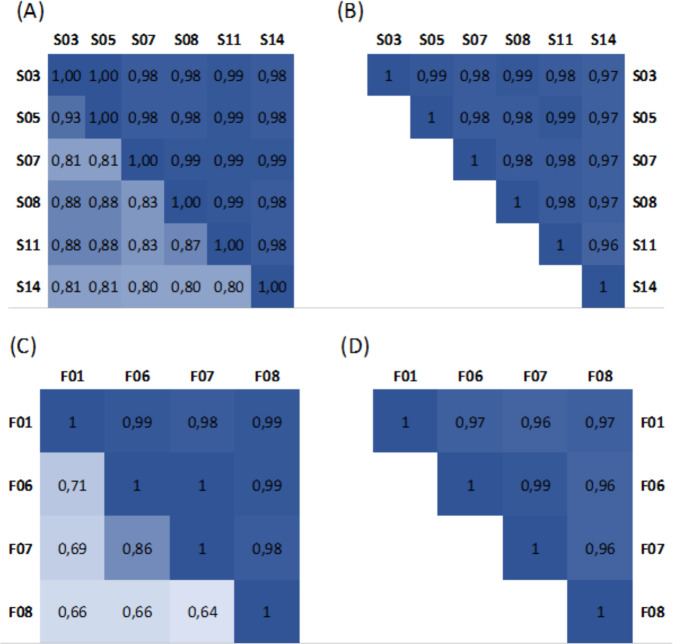
References
-
- Jansson JK, Baker ES. A multi-omic future for microbiome studies. Nat. Microbiol. 2016;1:16049. - PubMed
-
- Rodriguez-Valera F. Environmental genomics, the big picture? FEMS Microbiol. Lett. 2004;231:153–158. - PubMed
-
- Wilmes P, Bond PL. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004;6:911–920. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
