

Spatial genomics enables multi-modal study of clonal heterogeneity in tissues
- PMID: 34912115
- PMCID: PMC9301586
- DOI: 10.1038/s41586-021-04217-4
Spatial genomics enables multi-modal study of clonal heterogeneity in tissues
Abstract
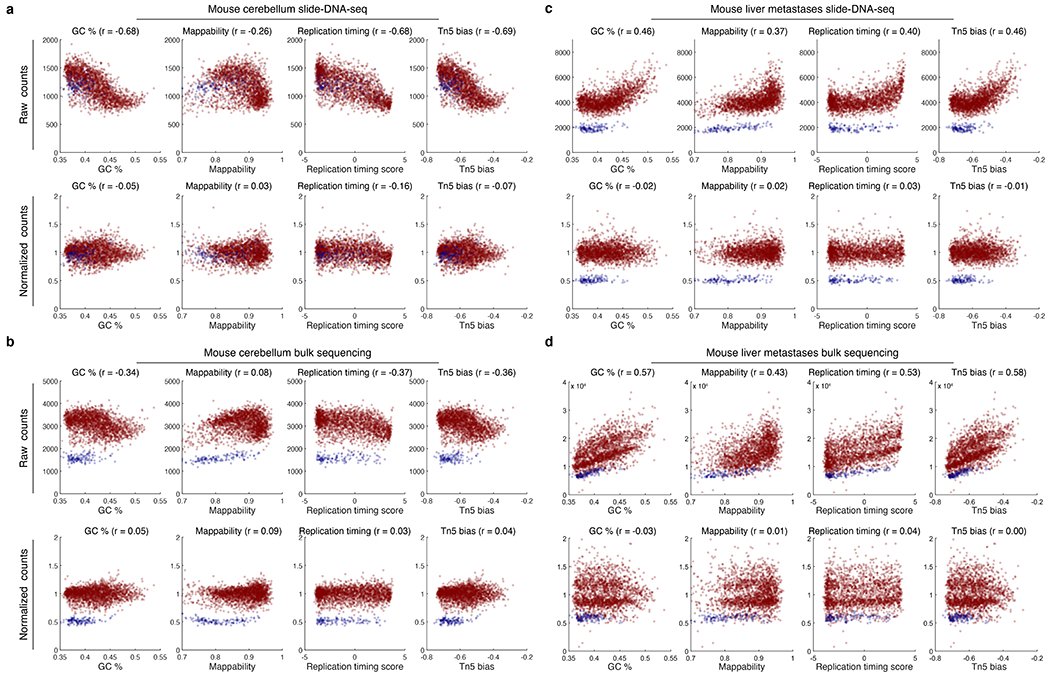
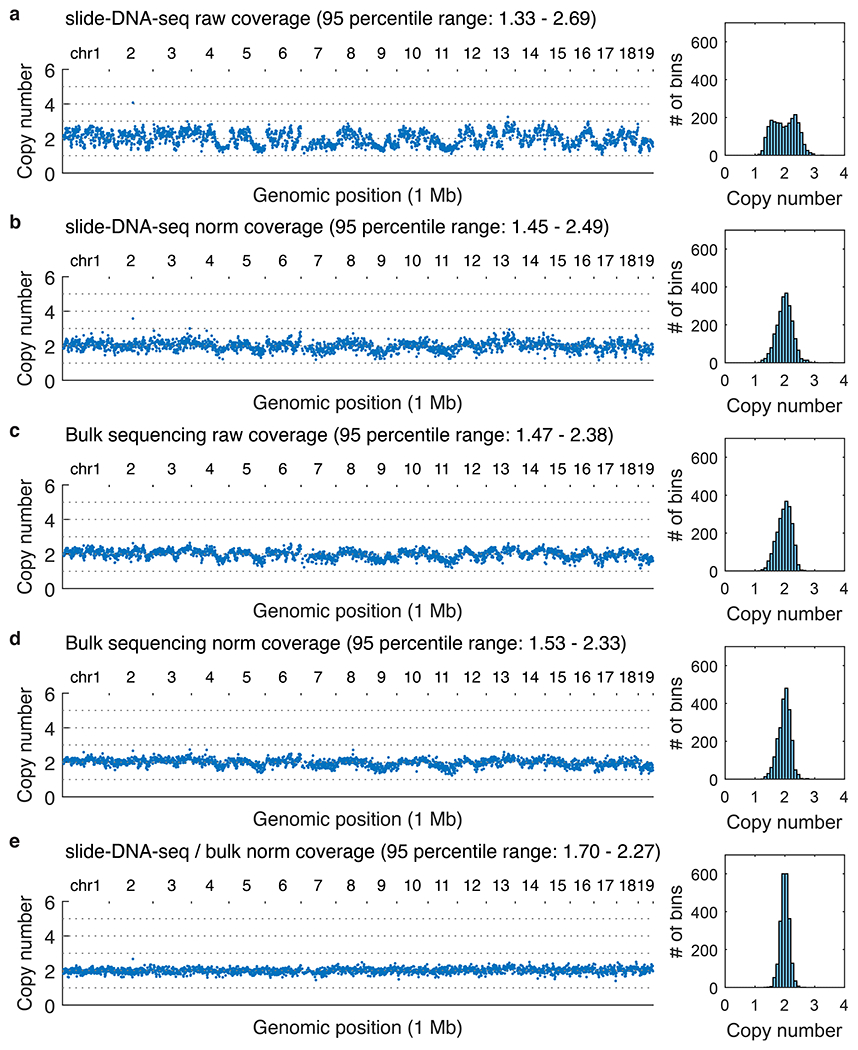
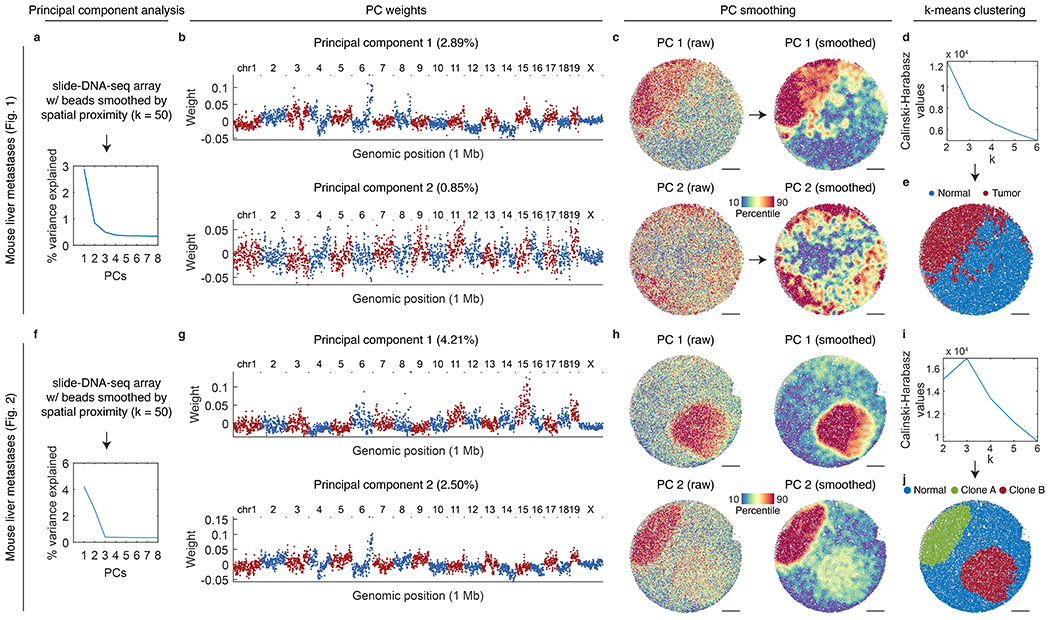
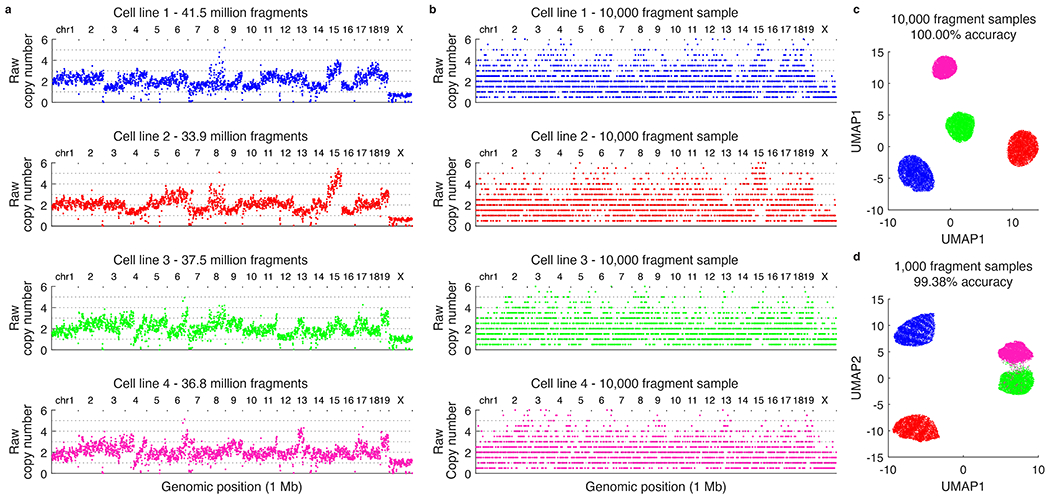
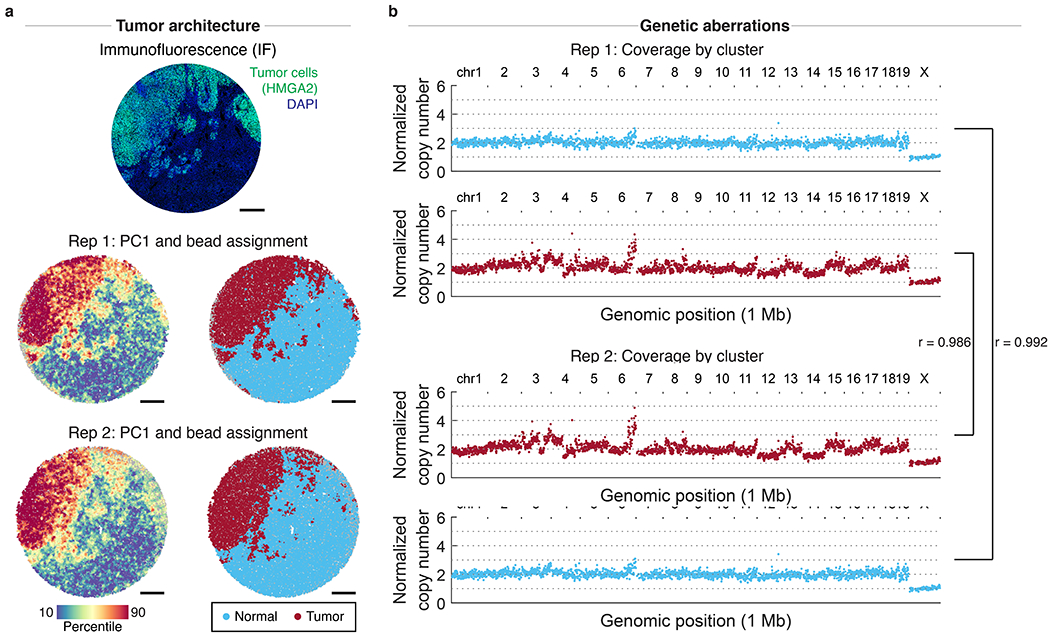
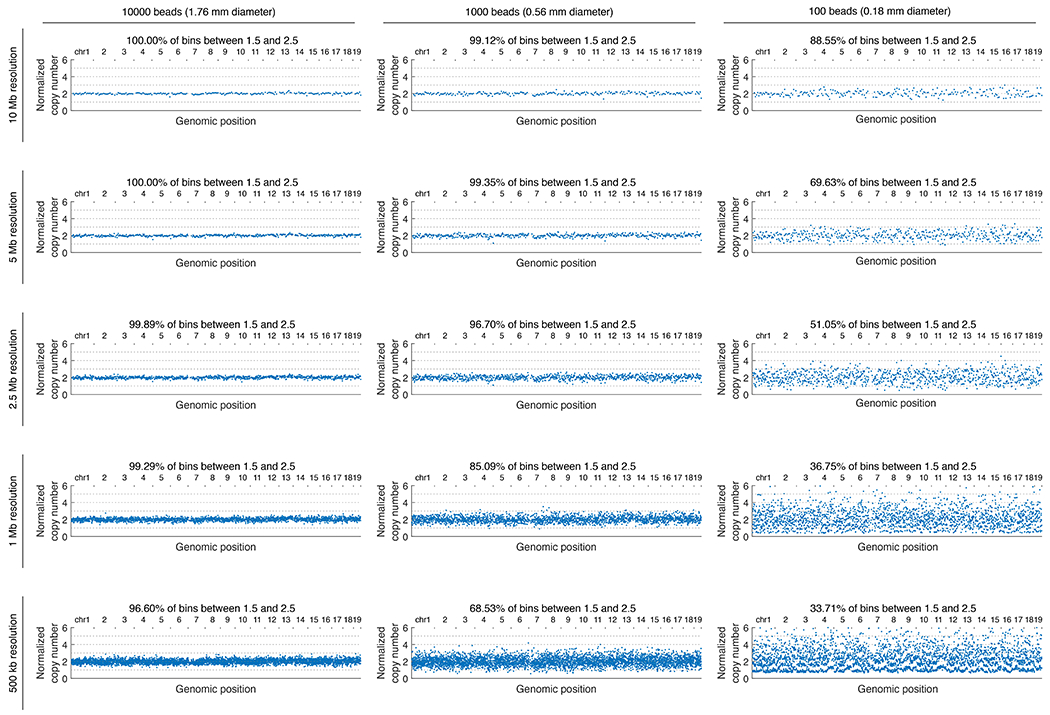
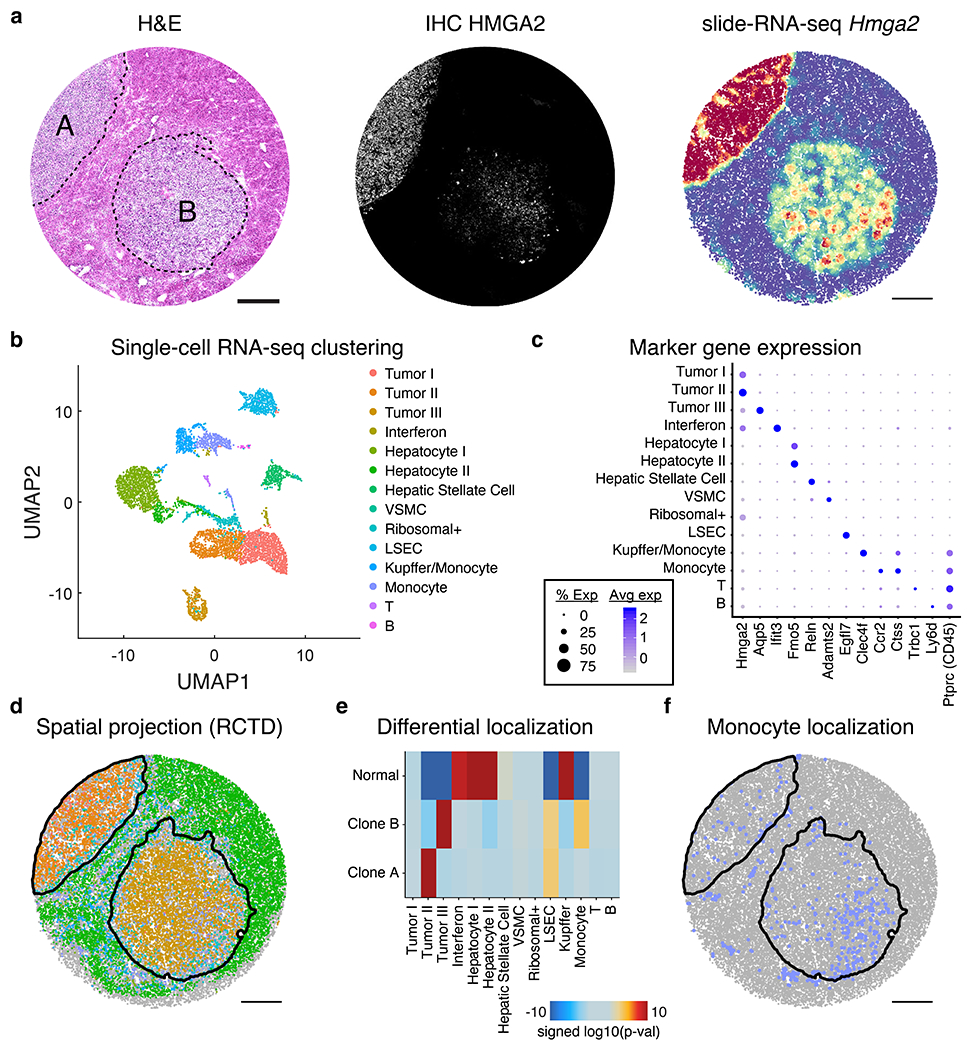
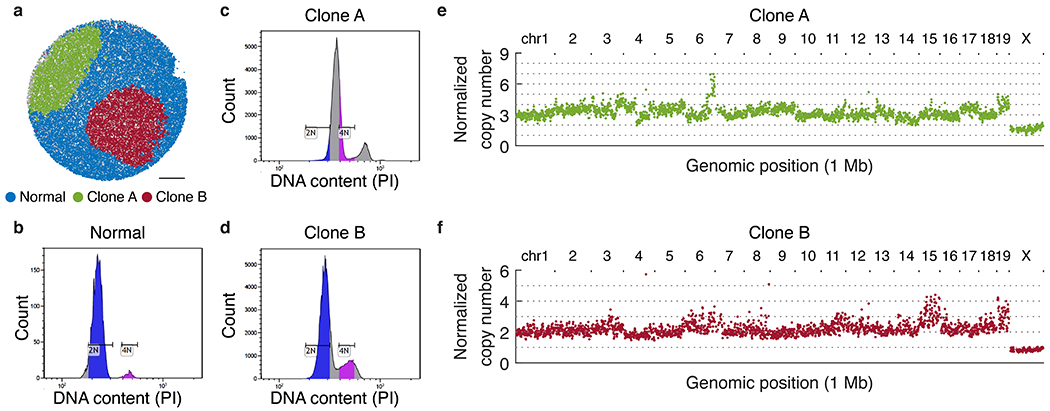
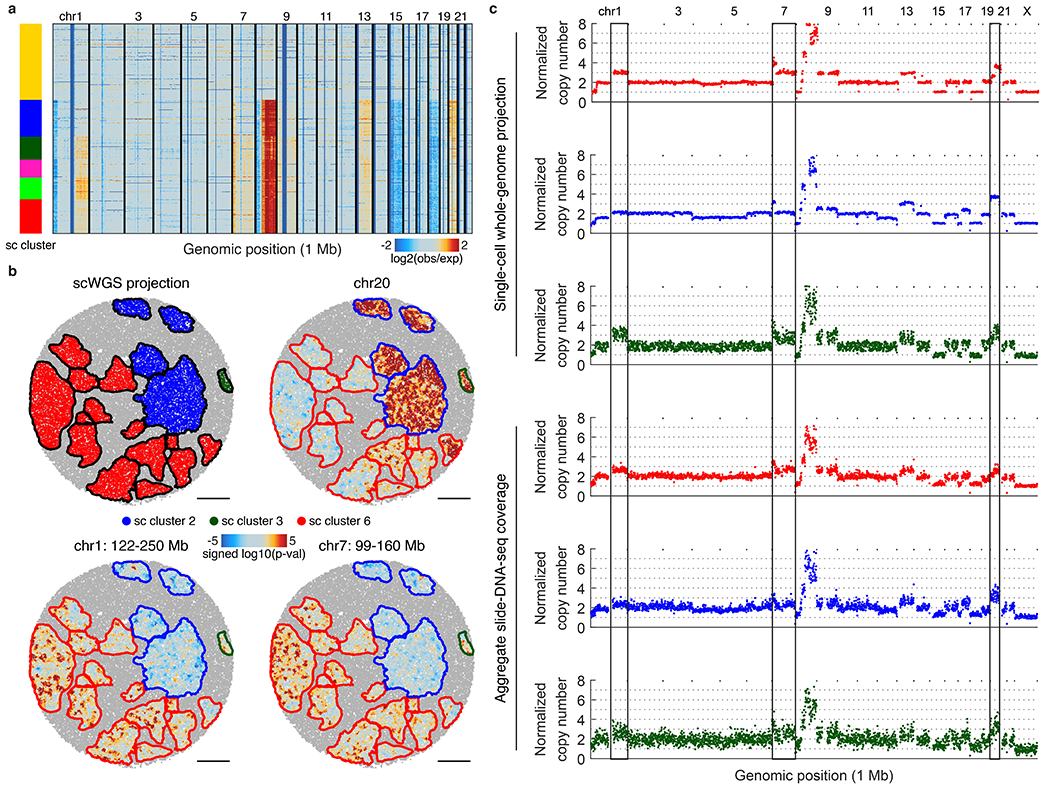
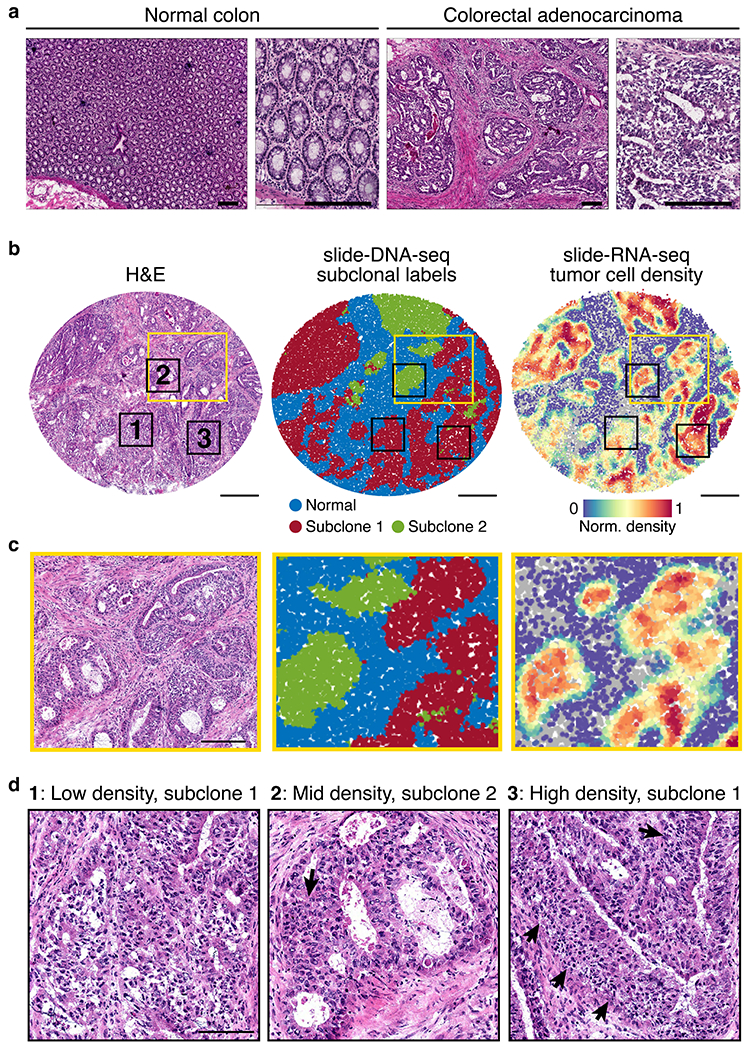
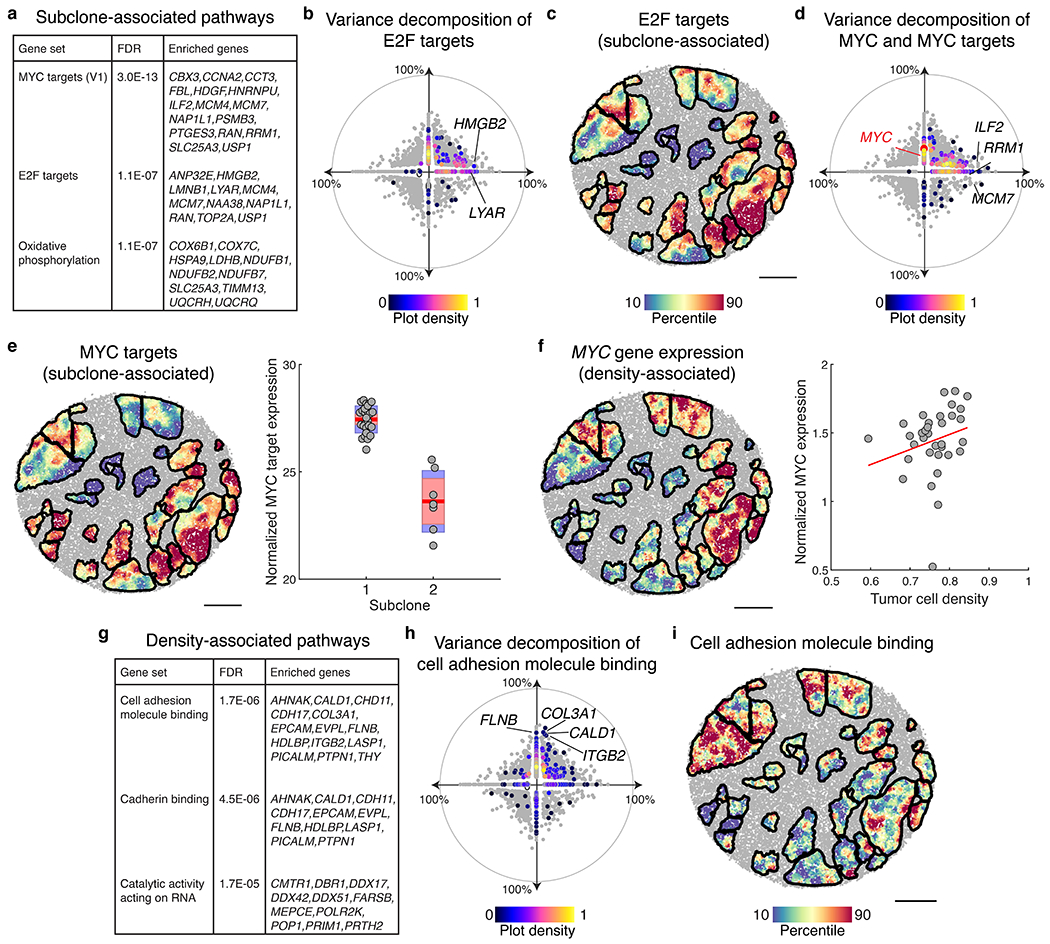
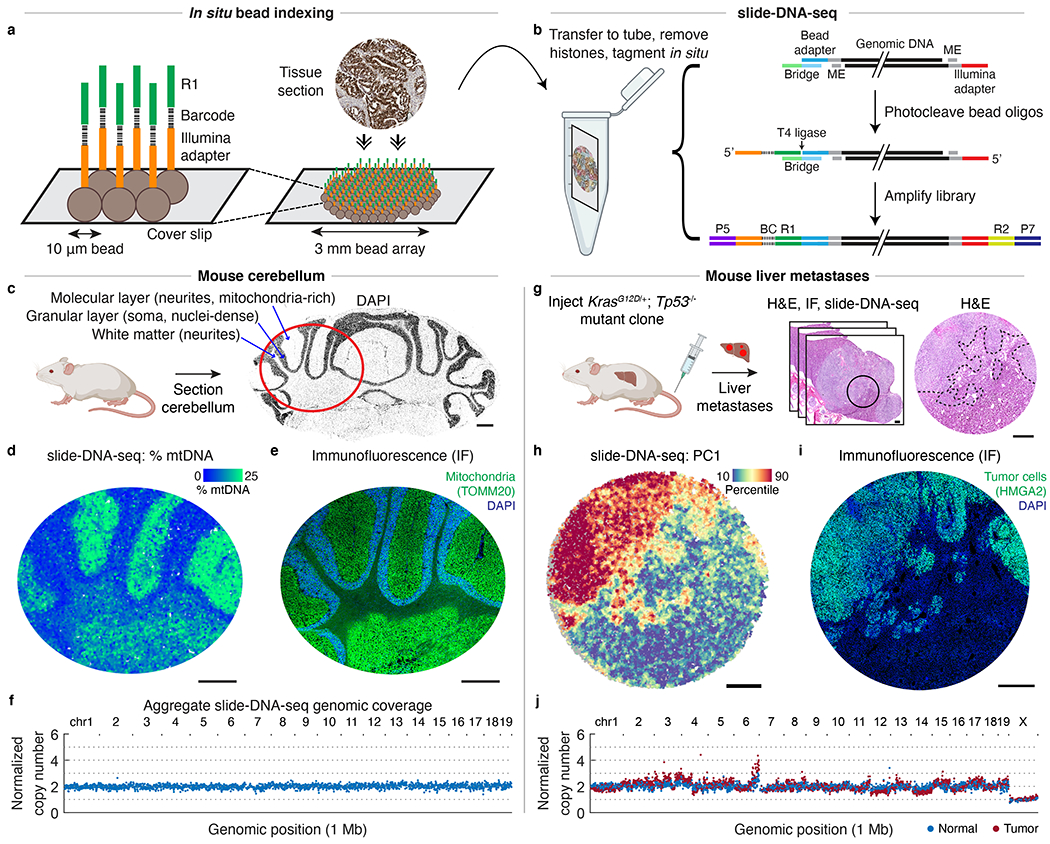
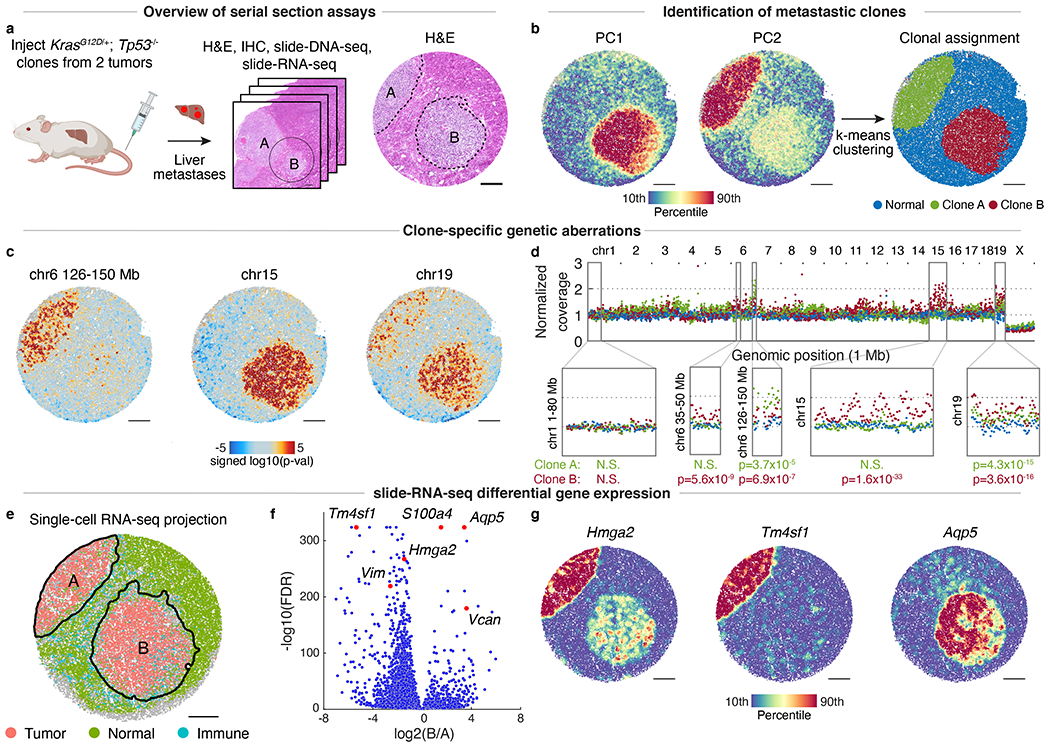
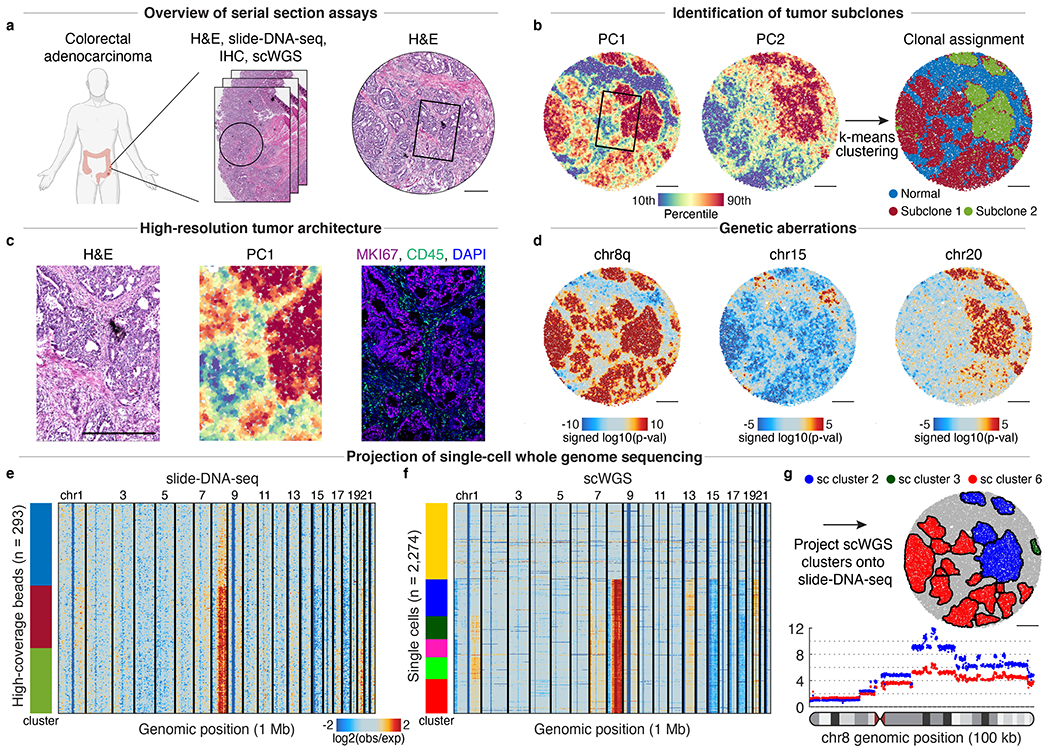
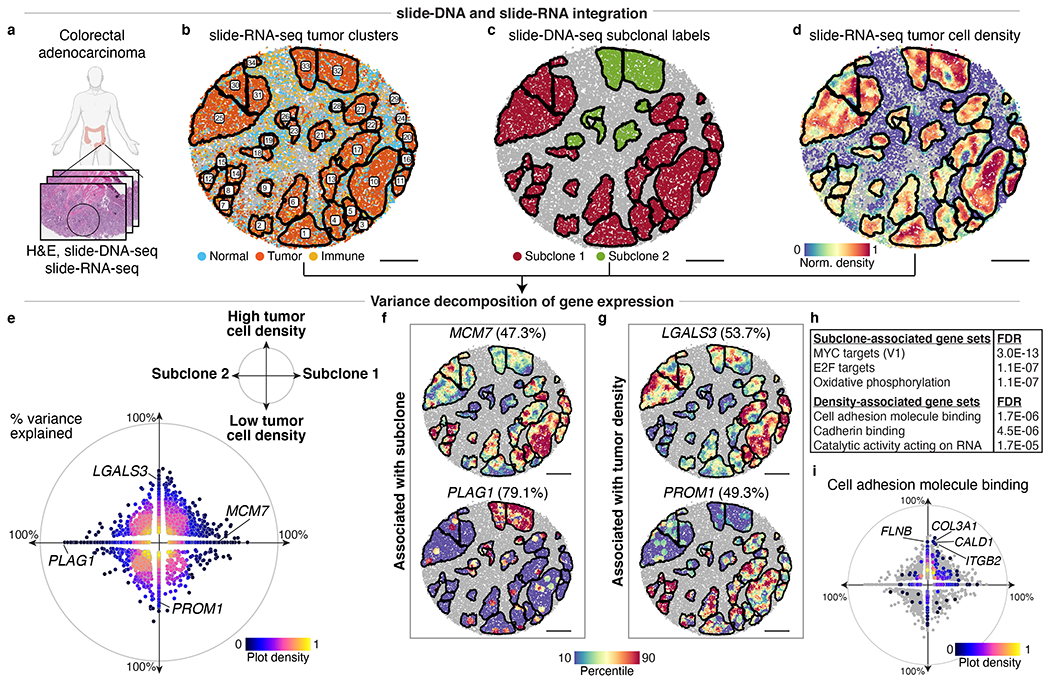
The state and behaviour of a cell can be influenced by both genetic and environmental factors. In particular, tumour progression is determined by underlying genetic aberrations1-4 as well as the makeup of the tumour microenvironment5,6. Quantifying the contributions of these factors requires new technologies that can accurately measure the spatial location of genomic sequence together with phenotypic readouts. Here we developed slide-DNA-seq, a method for capturing spatially resolved DNA sequences from intact tissue sections. We demonstrate that this method accurately preserves local tumour architecture and enables the de novo discovery of distinct tumour clones and their copy number alterations. We then apply slide-DNA-seq to a mouse model of metastasis and a primary human cancer, revealing that clonal populations are confined to distinct spatial regions. Moreover, through integration with spatial transcriptomics, we uncover distinct sets of genes that are associated with clone-specific genetic aberrations, the local tumour microenvironment, or both. Together, this multi-modal spatial genomics approach provides a versatile platform for quantifying how cell-intrinsic and cell-extrinsic factors contribute to gene expression, protein abundance and other cellular phenotypes.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
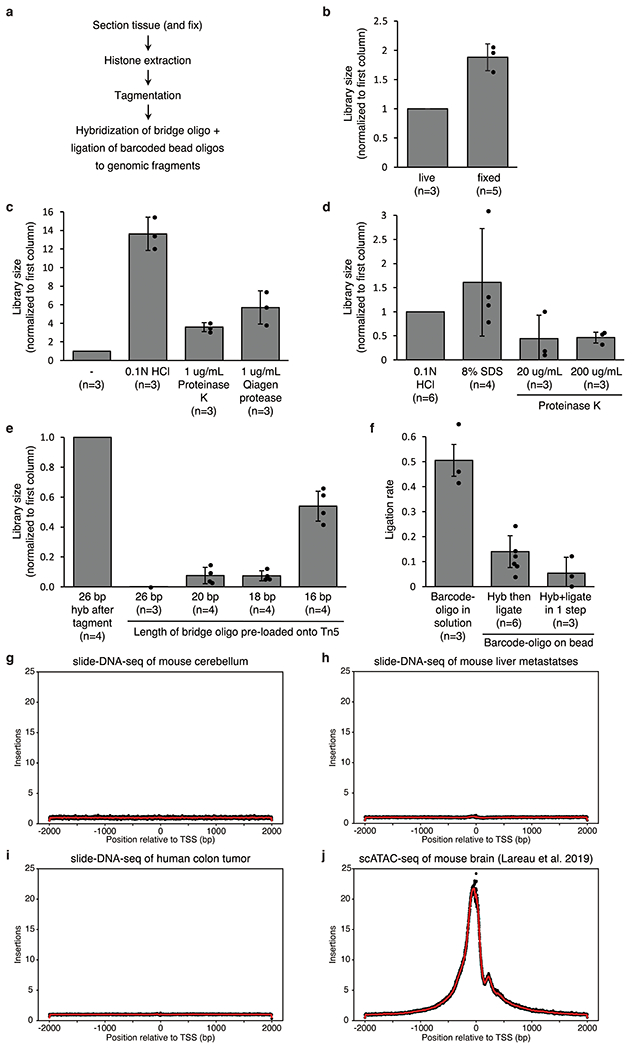
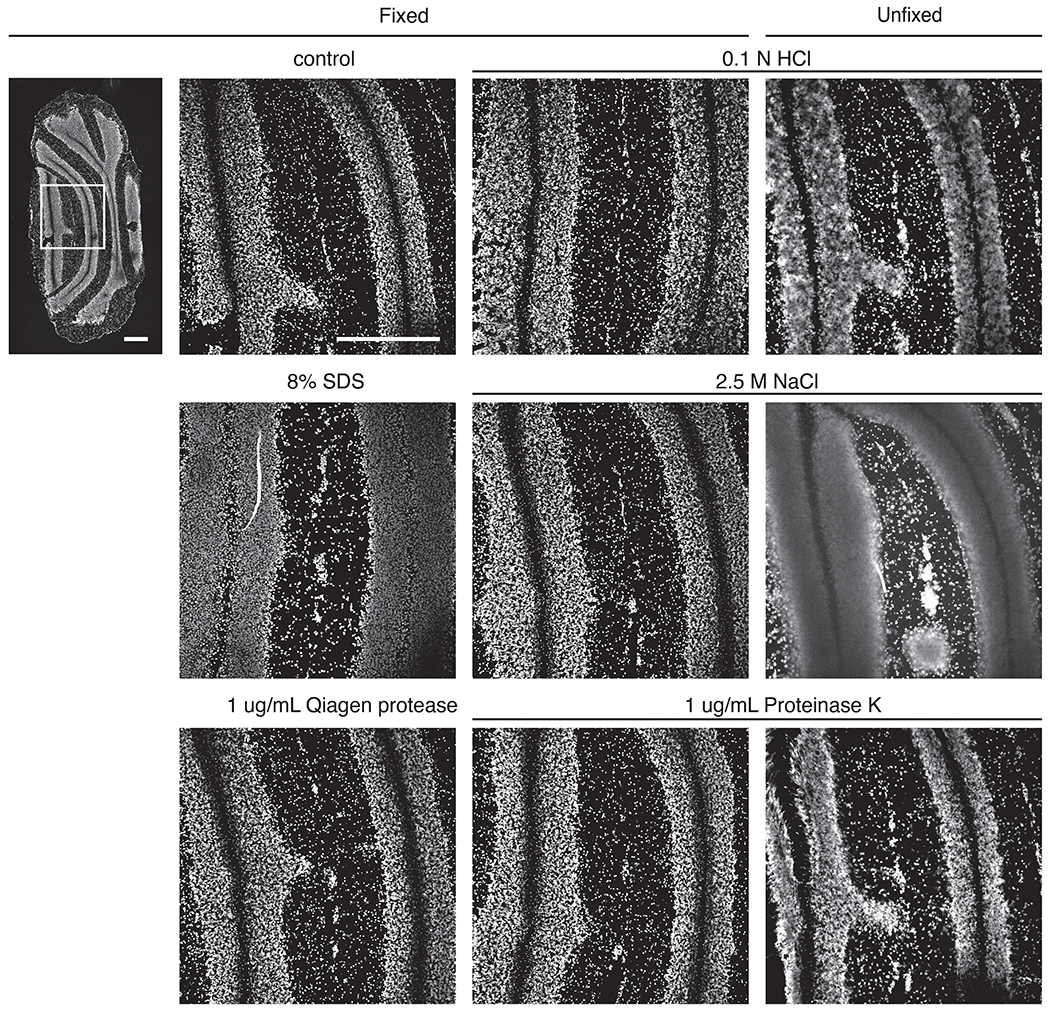
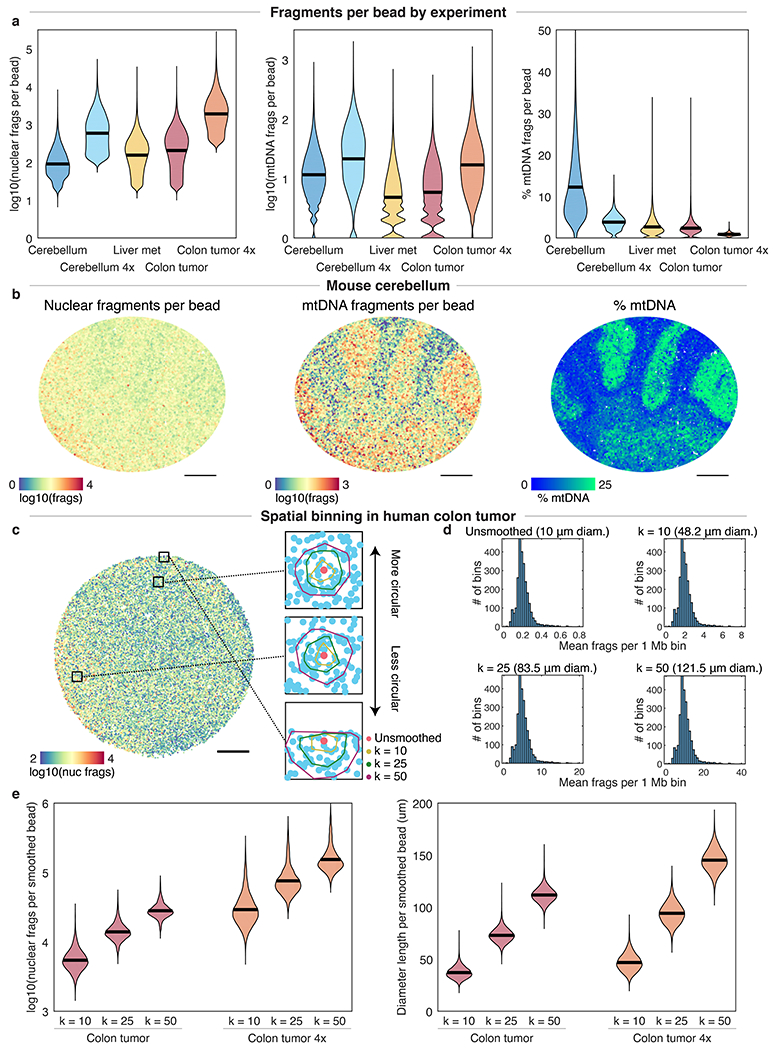
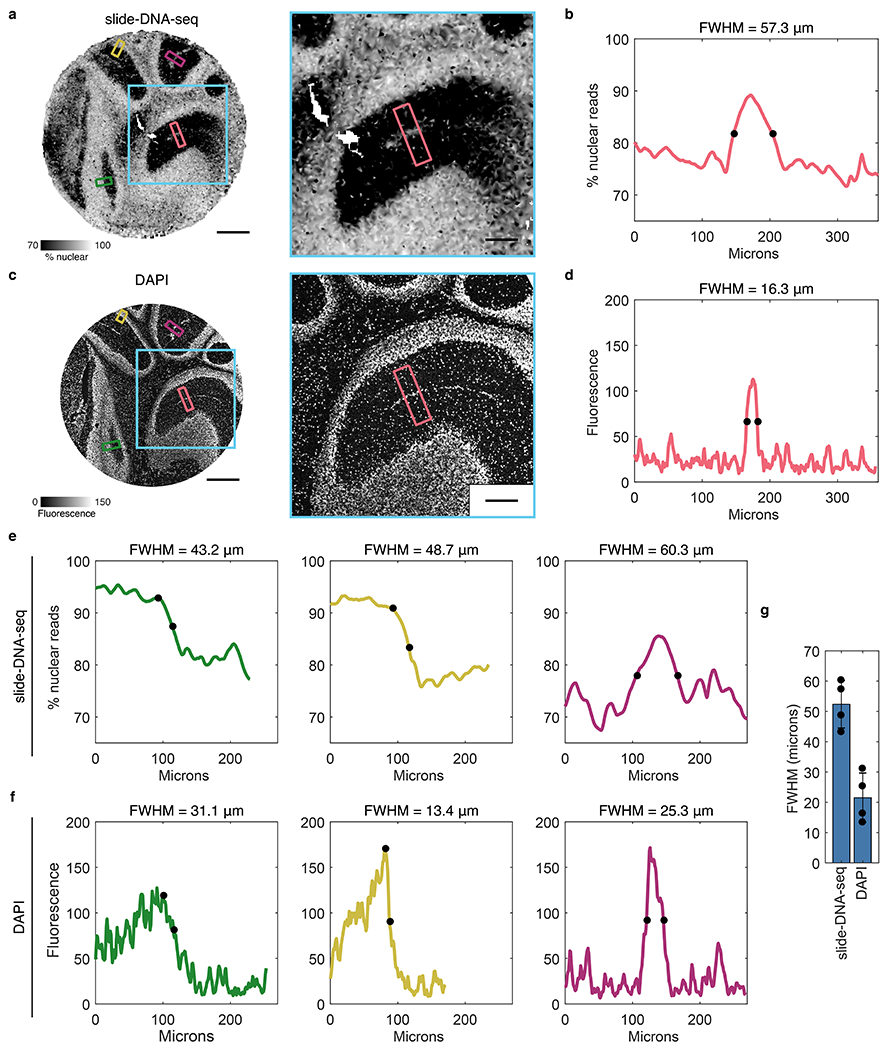
Comment in
-
Spatially resolved DNA sequencing.Nat Methods. 2022 Feb;19(2):139. doi: 10.1038/s41592-022-01405-6. Nat Methods. 2022. PMID: 35145316 No abstract available.
References
-
- McGranahan N & Swanton C Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628 (2017). - PubMed
-
- Turajlic S, Sottoriva A, Graham T & Swanton C Resolving genetic heterogeneity in cancer. Nat. Rev. Genet 20, 404–416 (2019). - PubMed
-
- Duan Q, Zhang H, Zheng J & Zhang L Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer Res. 6, 605–618 (2020). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
