

Towards the biogeography of prokaryotic genes
- PMID: 34912116
- PMCID: PMC7613196
- DOI: 10.1038/s41586-021-04233-4
Towards the biogeography of prokaryotic genes
Abstract
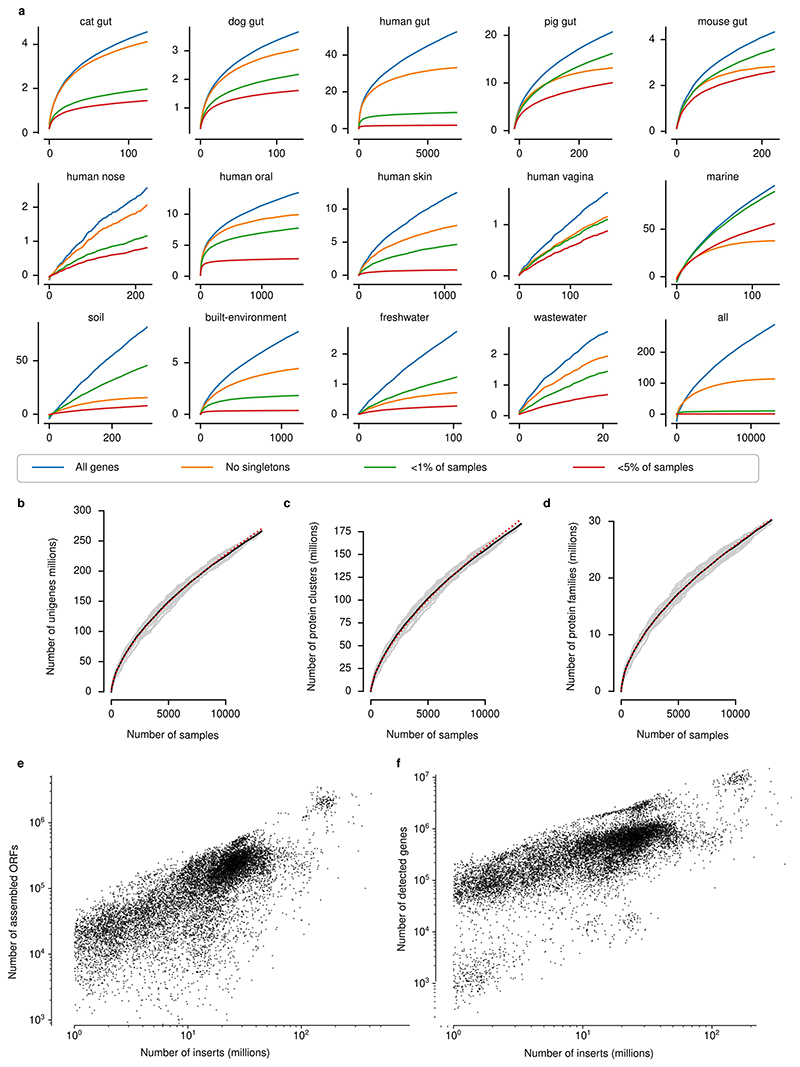
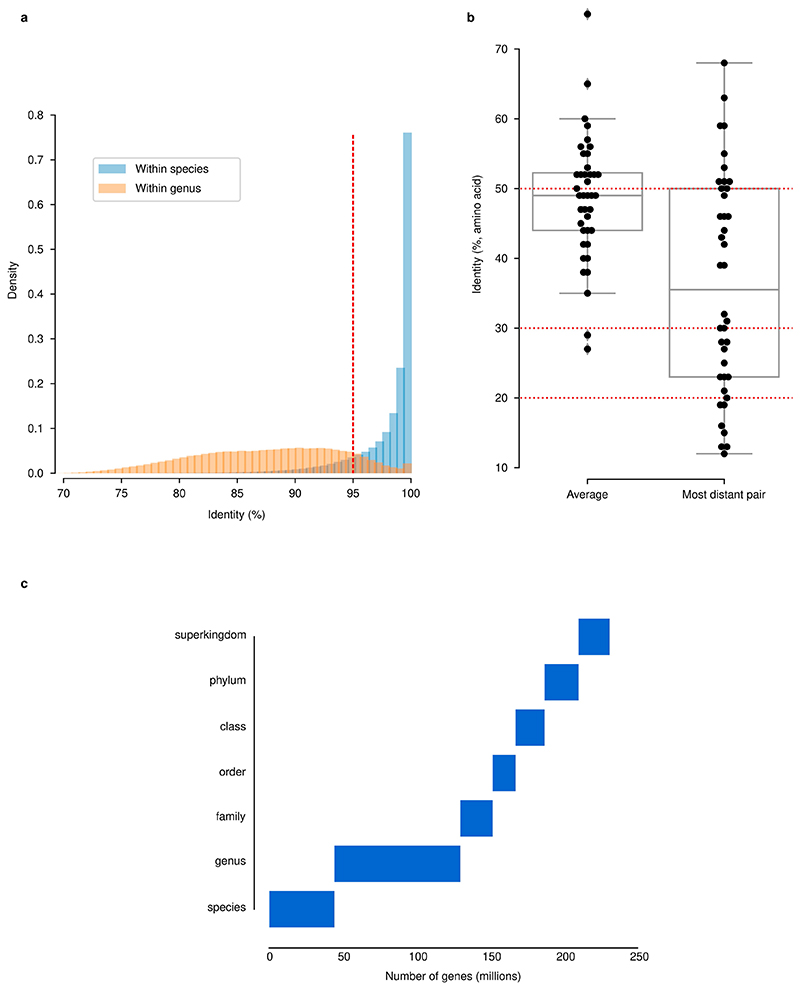
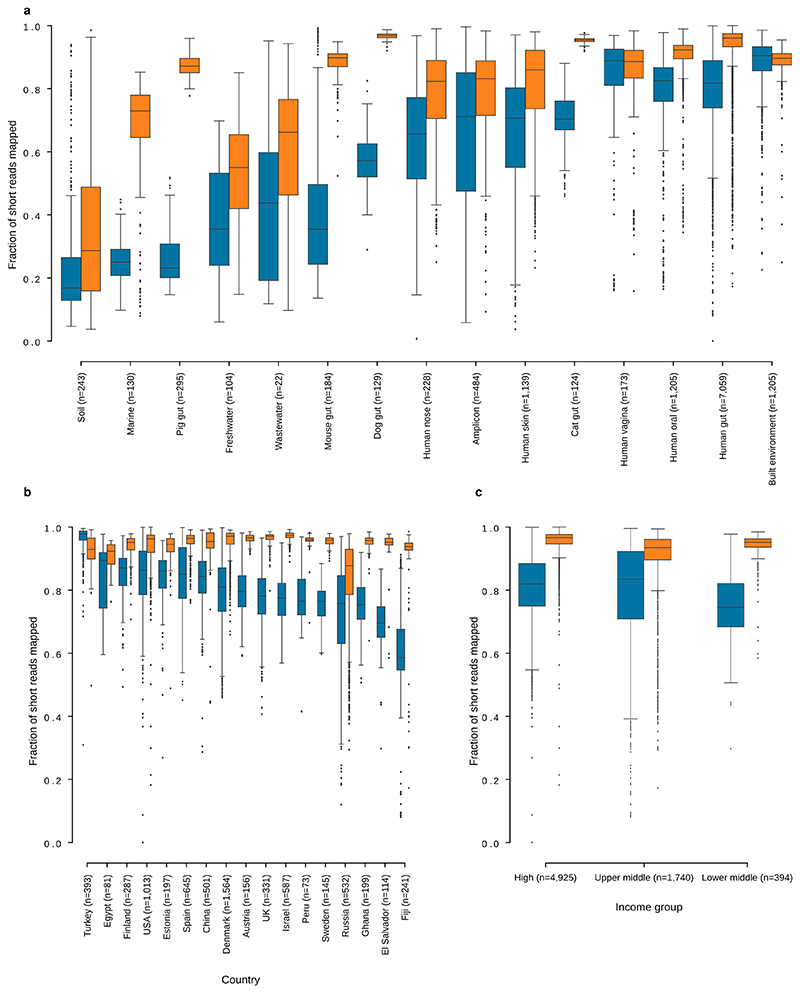
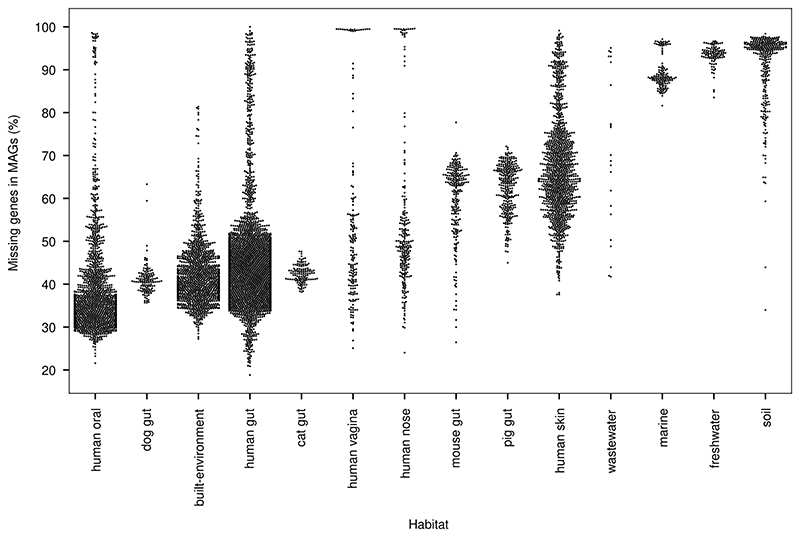
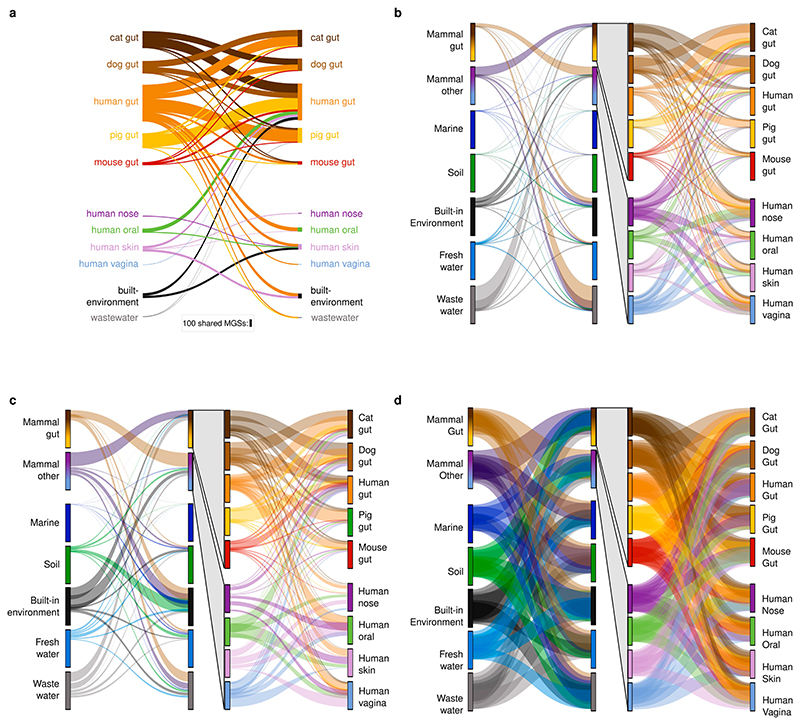
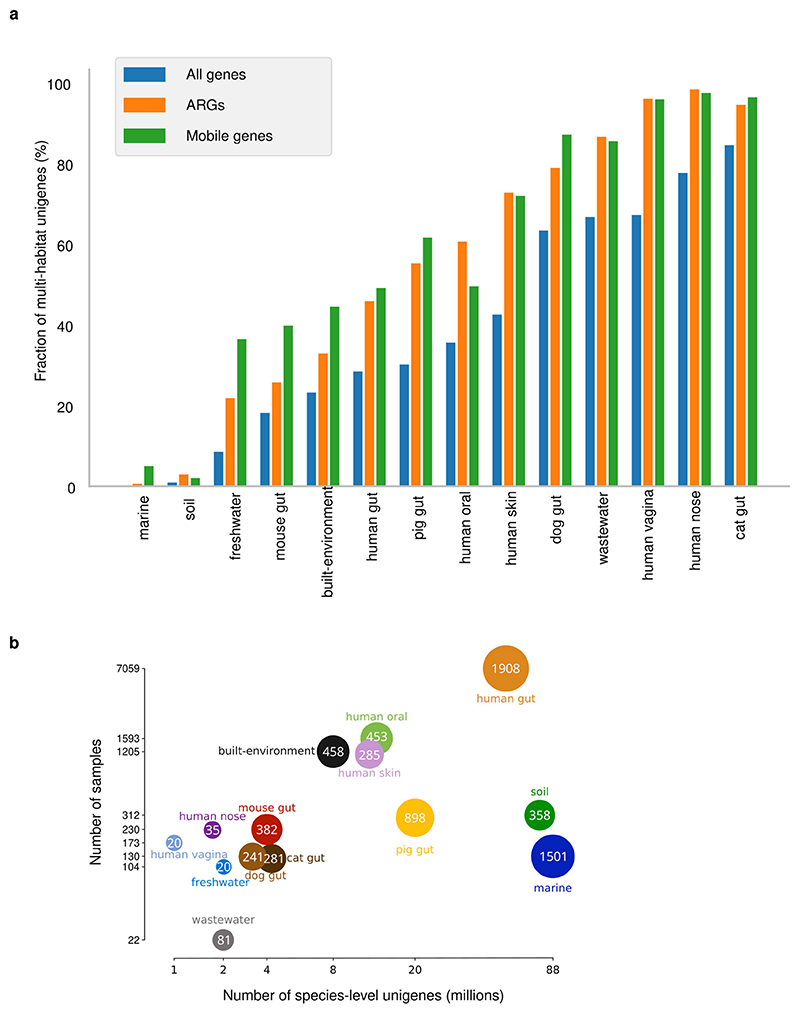
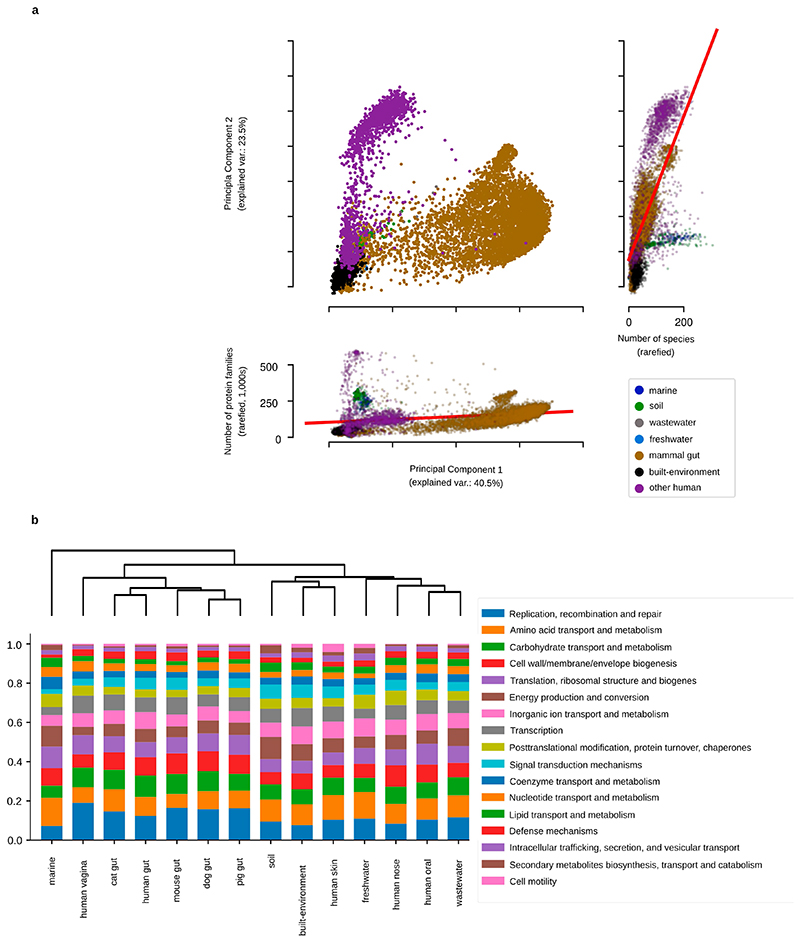
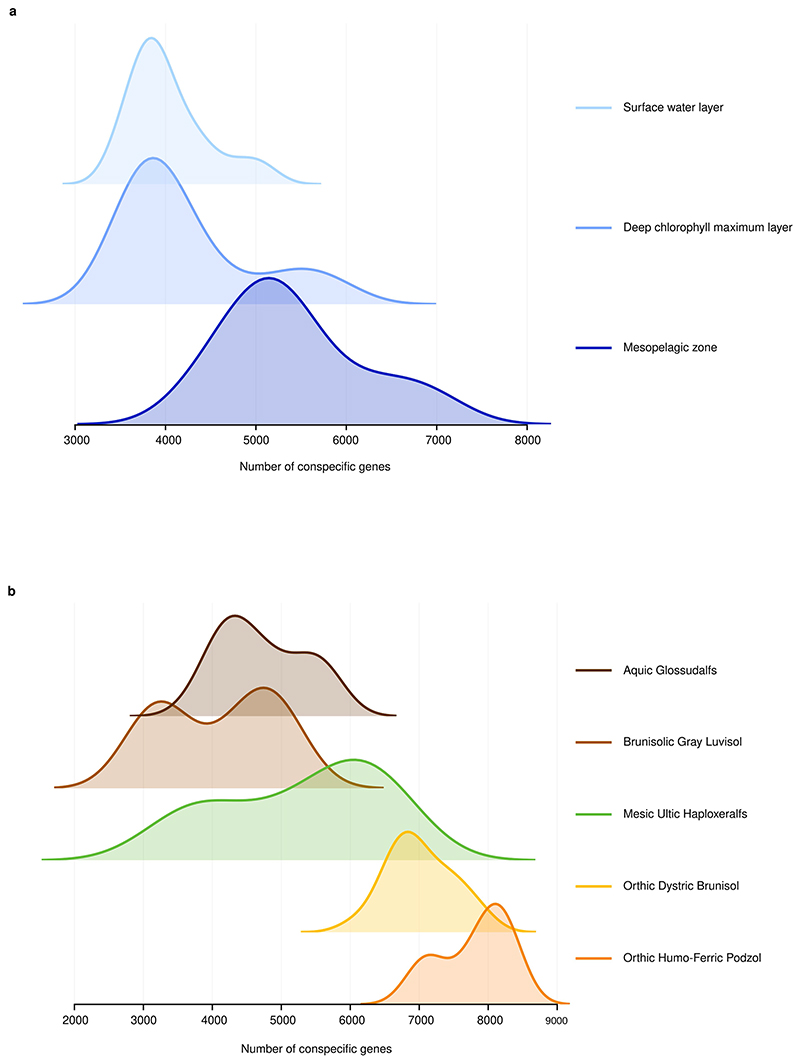
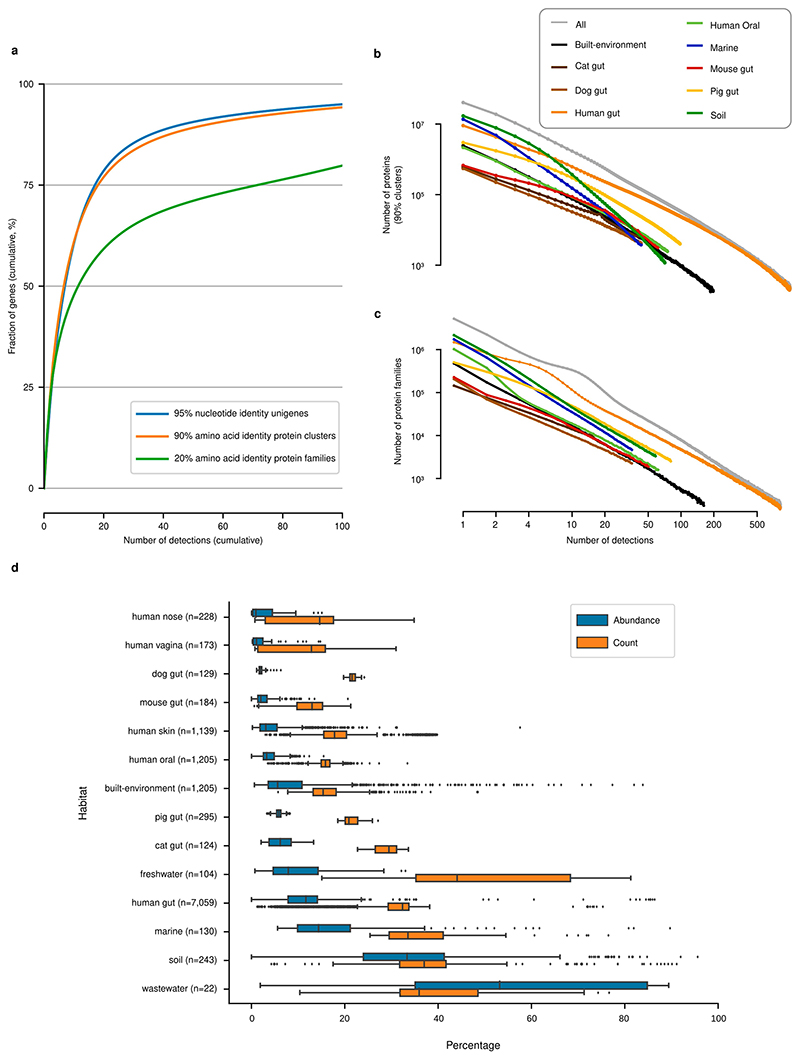
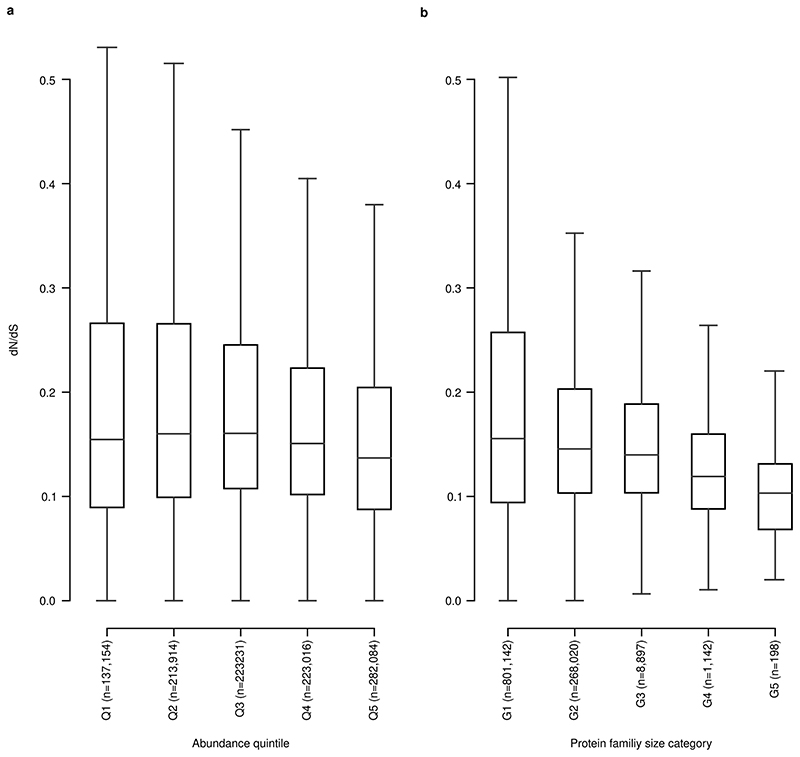
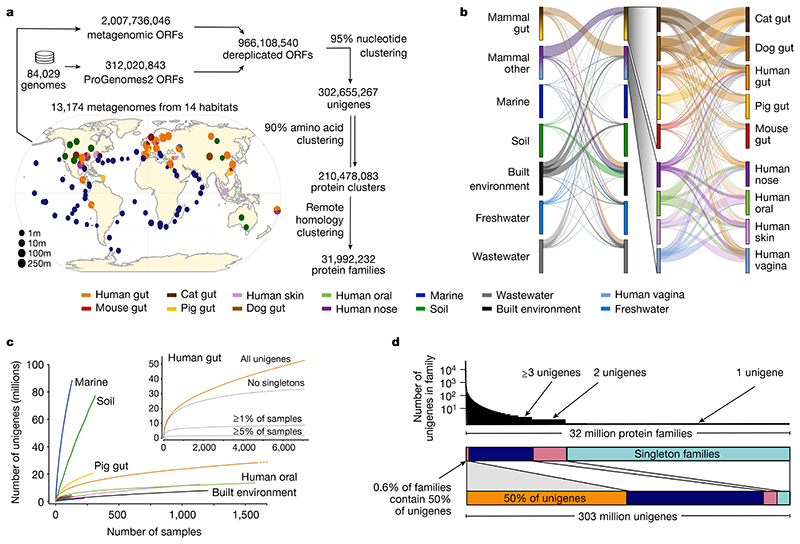
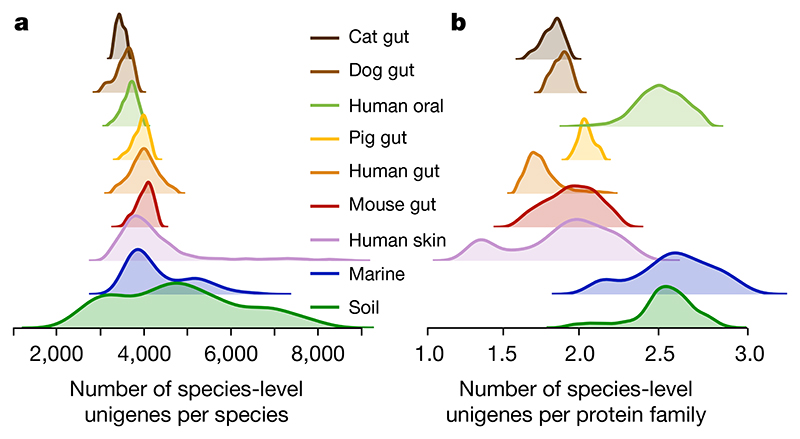
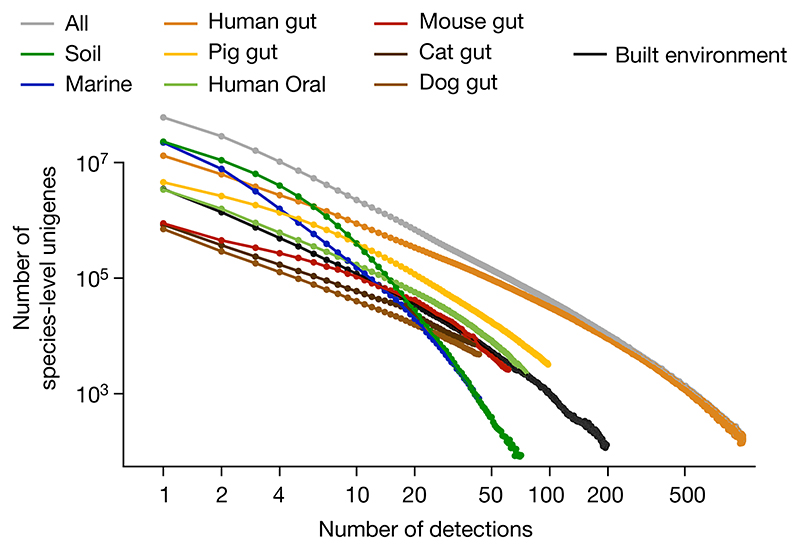
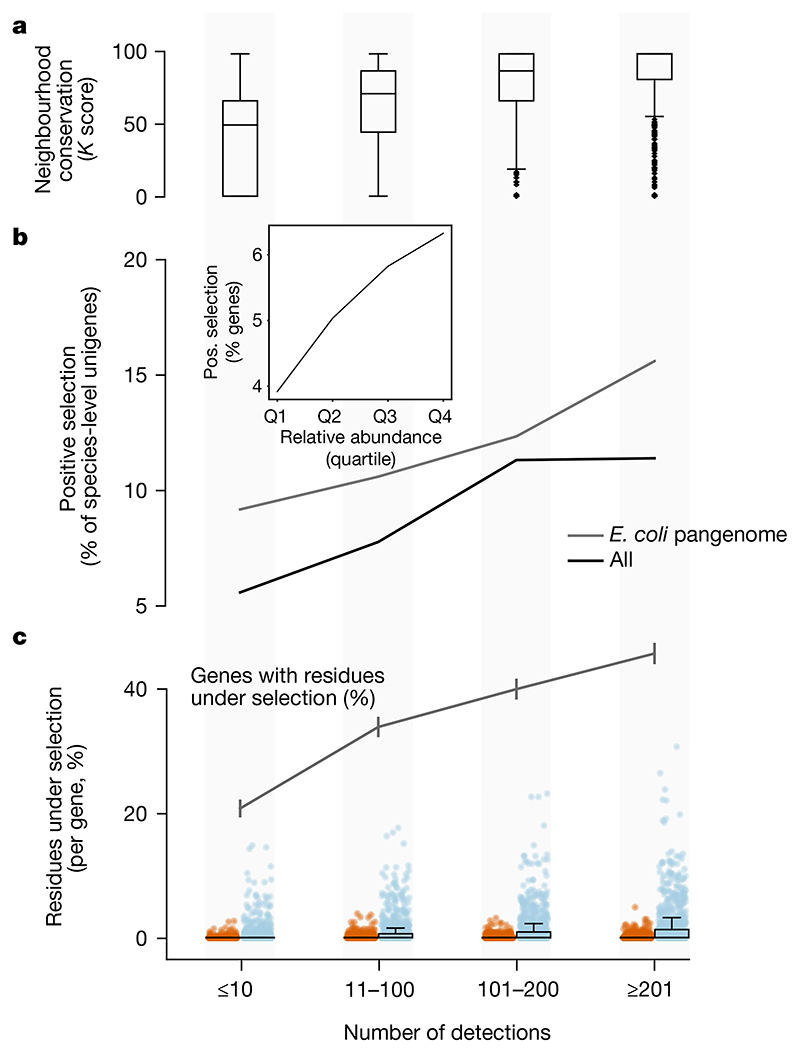
Microbial genes encode the majority of the functional repertoire of life on earth. However, despite increasing efforts in metagenomic sequencing of various habitats1-3, little is known about the distribution of genes across the global biosphere, with implications for human and planetary health. Here we constructed a non-redundant gene catalogue of 303 million species-level genes (clustered at 95% nucleotide identity) from 13,174 publicly available metagenomes across 14 major habitats and use it to show that most genes are specific to a single habitat. The small fraction of genes found in multiple habitats is enriched in antibiotic-resistance genes and markers for mobile genetic elements. By further clustering these species-level genes into 32 million protein families, we observed that a small fraction of these families contain the majority of the genes (0.6% of families account for 50% of the genes). The majority of species-level genes and protein families are rare. Furthermore, species-level genes, and in particular the rare ones, show low rates of positive (adaptive) selection, supporting a model in which most genetic variability observed within each protein family is neutral or nearly neutral.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
References
-
- Sunagawa S, et al. Structure and function of the global ocean microbiome. Science. 2015;348:1261359. - PubMed
-
- Mohammad BF, et al. Structure and function of the global topsoil microbiome. Nature. 2018;560:233–237. - PubMed
-
- Xiao L, et al. A catalog of the mouse gut metagenome. Nat Biotechnol. 2015;33:1103–1108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
