

Pangenomics enables genotyping of known structural variants in 5202 diverse genomes
- PMID: 34914532
- PMCID: PMC9365333
- DOI: 10.1126/science.abg8871
Pangenomics enables genotyping of known structural variants in 5202 diverse genomes
Abstract
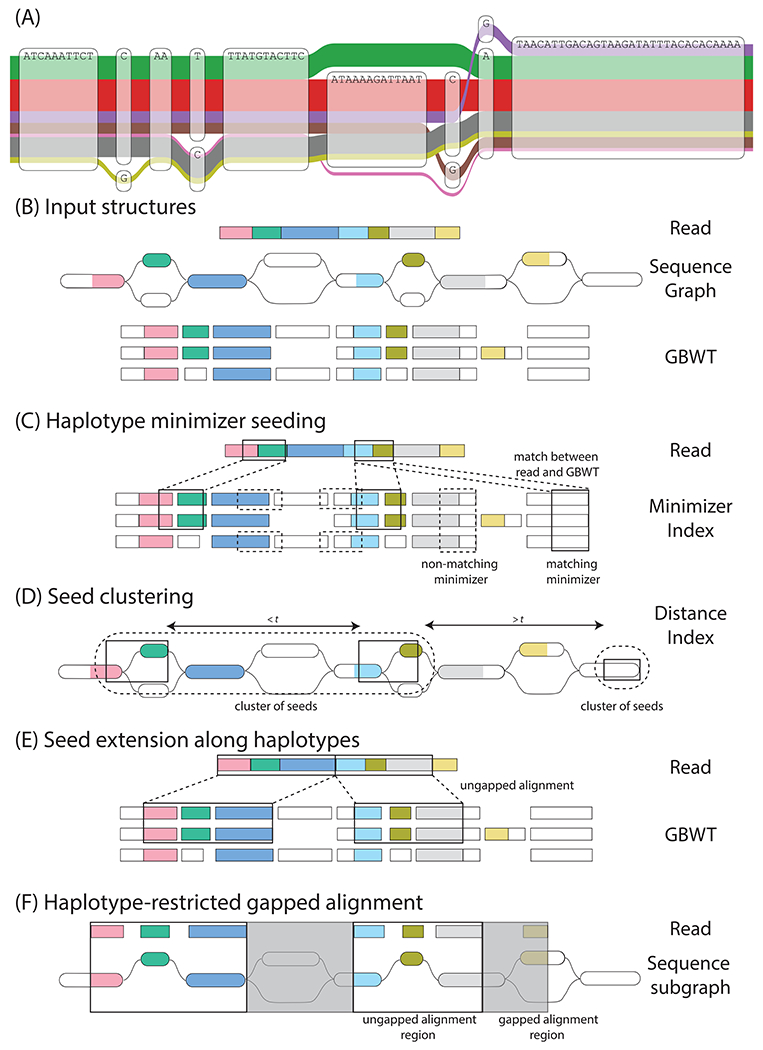
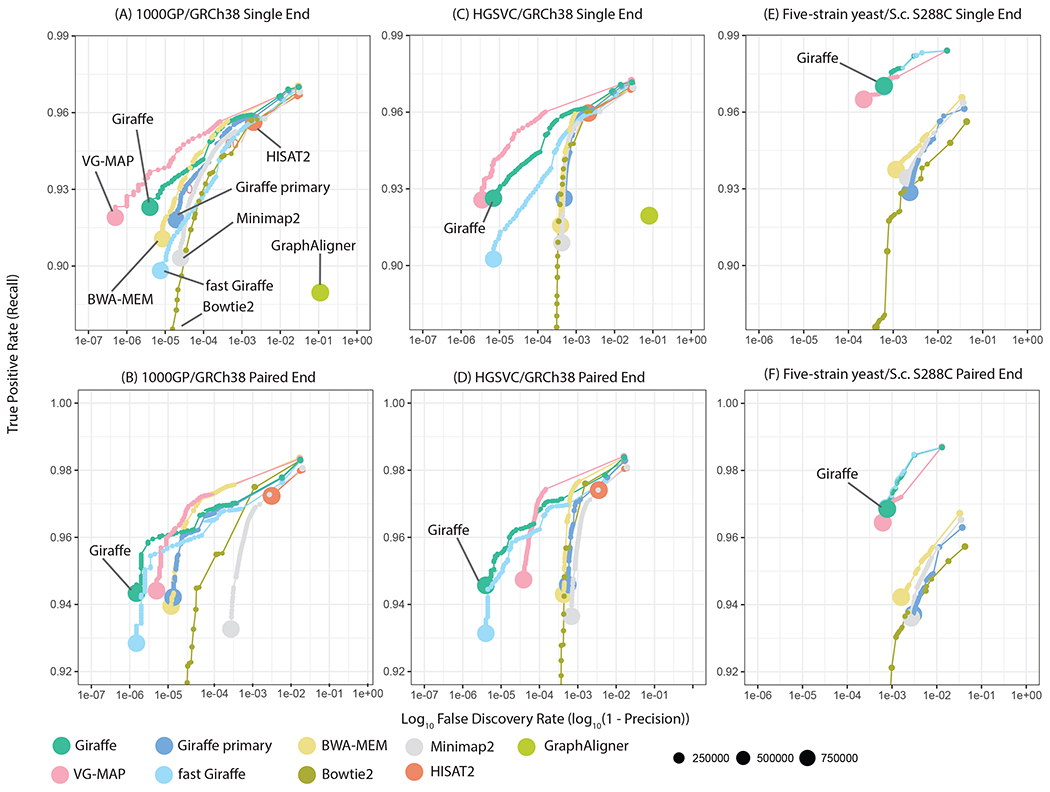
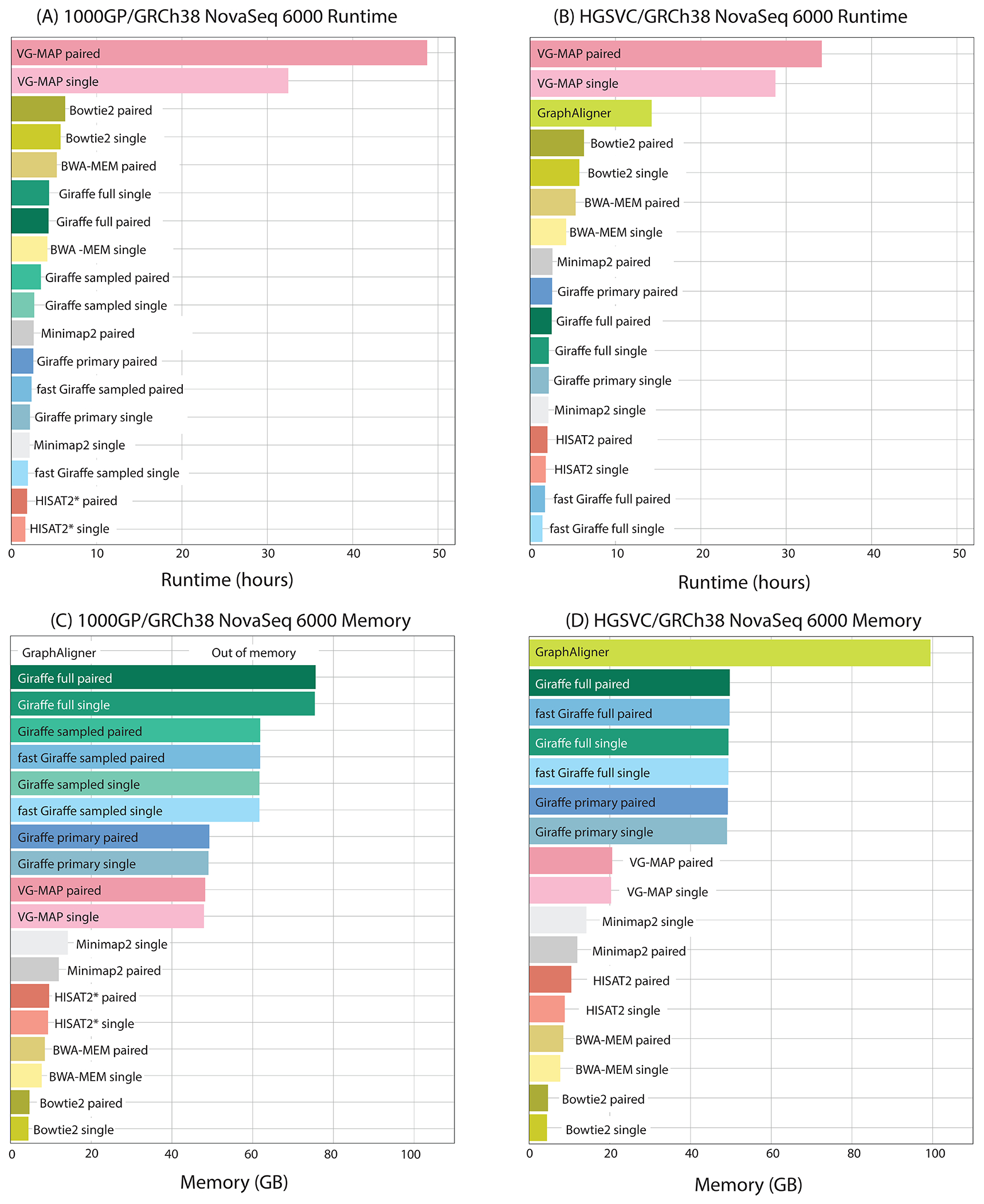
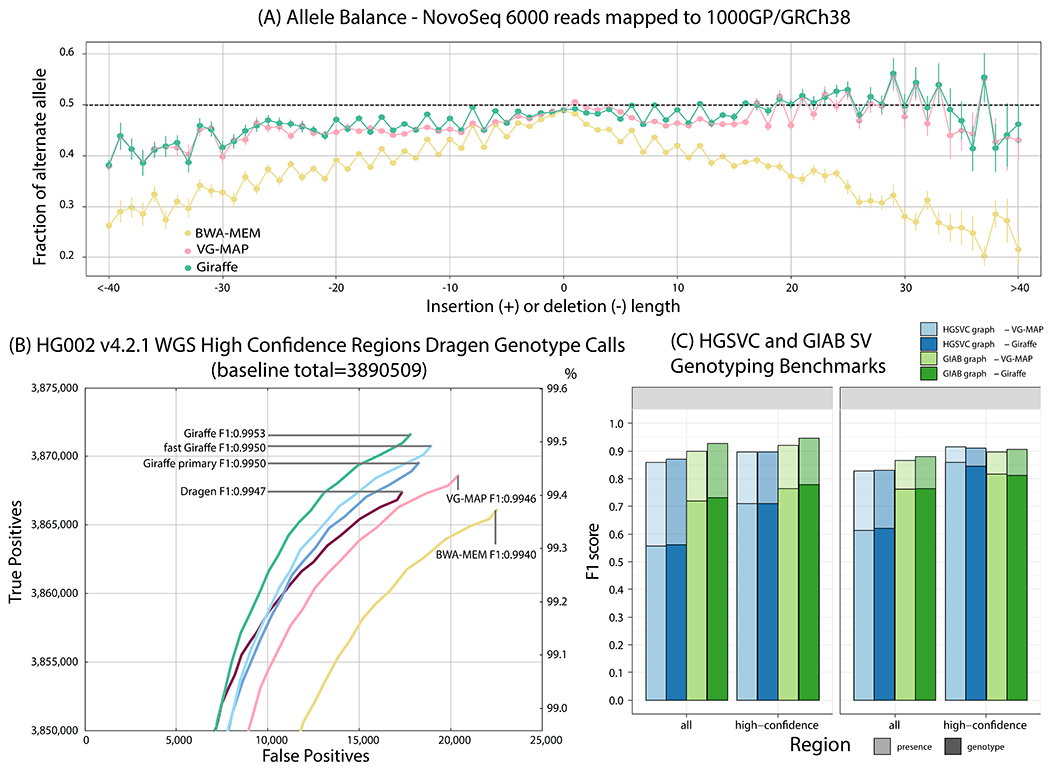
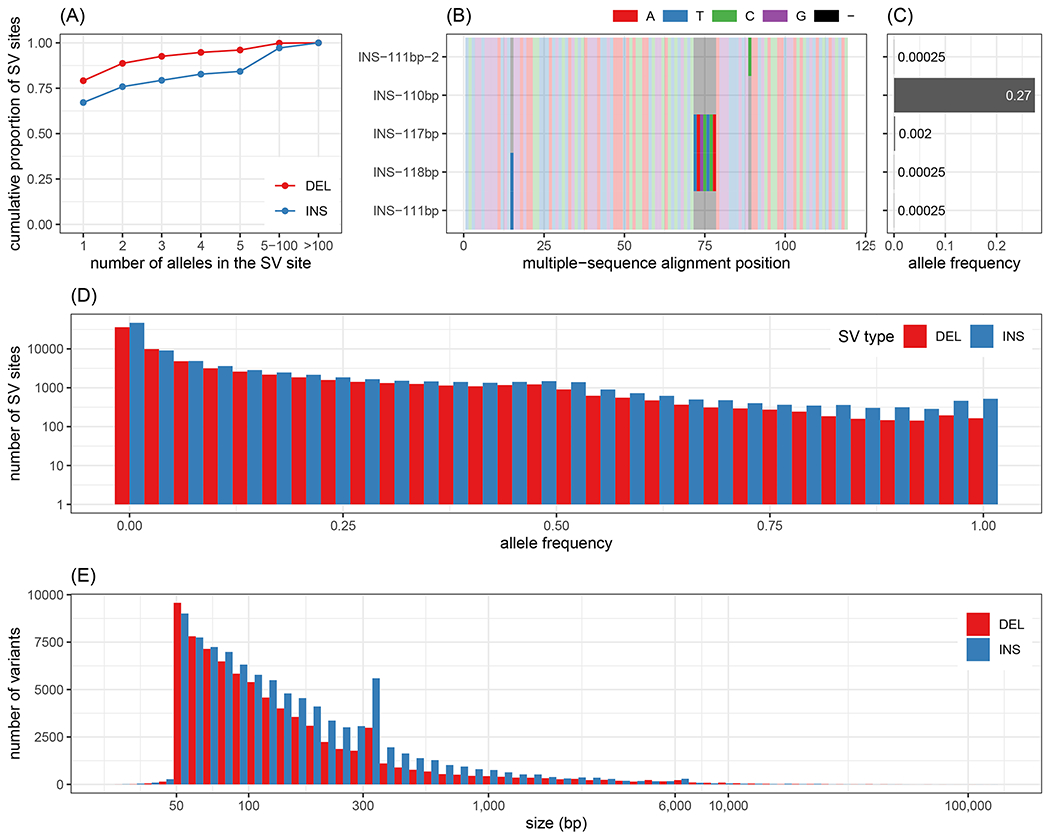
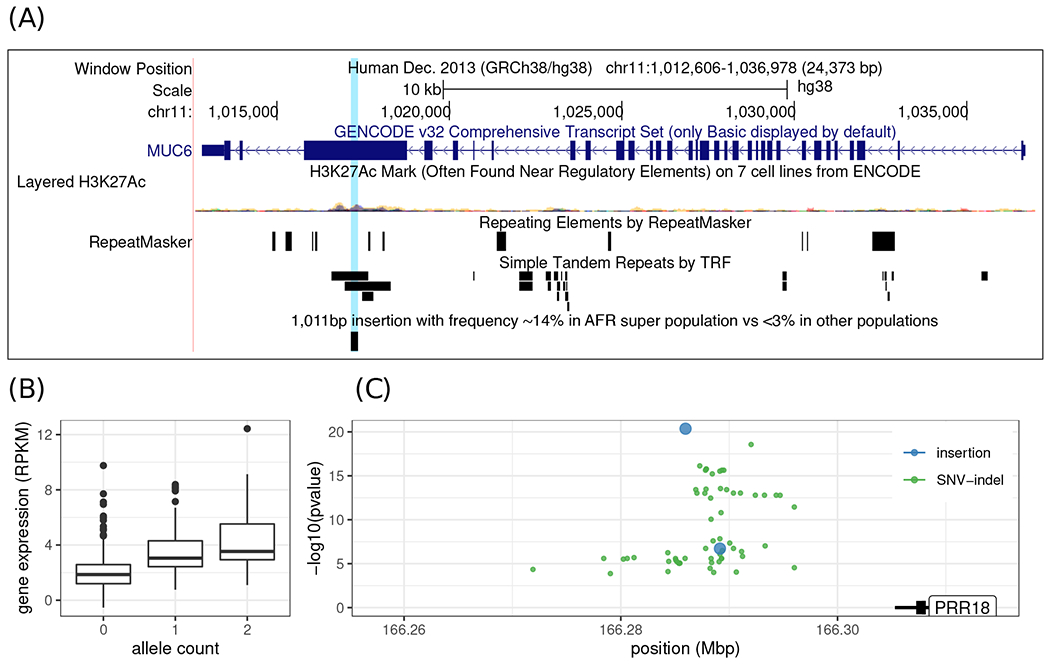
We introduce Giraffe, a pangenome short-read mapper that can efficiently map to a collection of haplotypes threaded through a sequence graph. Giraffe maps sequencing reads to thousands of human genomes at a speed comparable to that of standard methods mapping to a single reference genome. The increased mapping accuracy enables downstream improvements in genome-wide genotyping pipelines for both small variants and larger structural variants. We used Giraffe to genotype 167,000 structural variants, discovered in long-read studies, in 5202 diverse human genomes that were sequenced using short reads. We conclude that pangenomics facilitates a more comprehensive characterization of variation and, as a result, has the potential to improve many genomic analyses.
Conflict of interest statement
Competing interests
P.C. and A.C. are employees of Google and own Alphabet stock as part of the standard compensation package. The remaining authors declare no competing interests.
Figures
References
-
- Ebler J, Schönhuth A, Marschall T, Bioinformatics 33, 4015 (2017). - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
- U54 HG003067/HG/NHGRI NIH HHS/United States
- 75N92020D00001/HL/NHLBI NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- U24 HG007234/HG/NHGRI NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- UL1 TR000040/TR/NCATS NIH HHS/United States
- N01 HC095166/HL/NHLBI NIH HHS/United States
- N01 HC095160/HL/NHLBI NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- 75N92020D00002/HL/NHLBI NIH HHS/United States
- HHSN268201500003C/HL/NHLBI NIH HHS/United States
- U01 HL137183/HL/NHLBI NIH HHS/United States
- N01 HC095161/HL/NHLBI NIH HHS/United States
- 75N92020D00005/HL/NHLBI NIH HHS/United States
- N01 HC095168/HL/NHLBI NIH HHS/United States
- R01 HL120393/HL/NHLBI NIH HHS/United States
- UL1 TR001079/TR/NCATS NIH HHS/United States
- N02 HL064278/HL/NHLBI NIH HHS/United States
- N01 HC095169/HL/NHLBI NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- N01 HC095167/HL/NHLBI NIH HHS/United States
- UM1 HG008901/HG/NHGRI NIH HHS/United States
- OT2 OD026682/OD/NIH HHS/United States
- OT3 HL142481/HL/NHLBI NIH HHS/United States
- N01 HC095159/HL/NHLBI NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- OT3 HL147154/HL/NHLBI NIH HHS/United States
- 75N92020D00003/HL/NHLBI NIH HHS/United States
- UL1 TR001420/TR/NCATS NIH HHS/United States
- 75N92020D00004/HL/NHLBI NIH HHS/United States
- N01 HC095163/HL/NHLBI NIH HHS/United States
- 75N92020D00007/HL/NHLBI NIH HHS/United States
- HHSN268201500014C/HL/NHLBI NIH HHS/United States
- HHSN268201500003I/HL/NHLBI NIH HHS/United States
- 75N92020D00006/HL/NHLBI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- N01 HC095162/HL/NHLBI NIH HHS/United States
- UL1 TR001881/TR/NCATS NIH HHS/United States
- N01 HC095165/HL/NHLBI NIH HHS/United States
- N01 HC095164/HL/NHLBI NIH HHS/United States
