

Engineering an efficient and enantioselective enzyme for the Morita-Baylis-Hillman reaction
- PMID: 34916595
- PMCID: PMC7612480
- DOI: 10.1038/s41557-021-00833-9
Engineering an efficient and enantioselective enzyme for the Morita-Baylis-Hillman reaction
Abstract
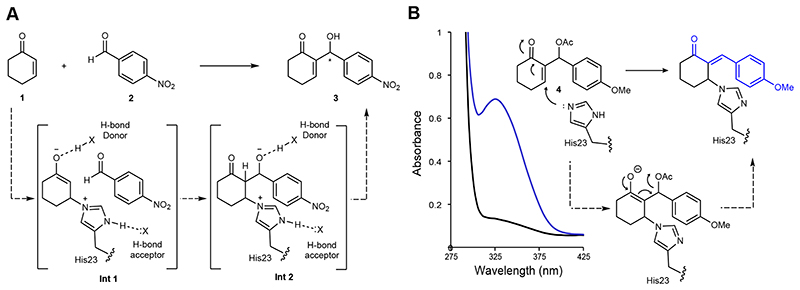
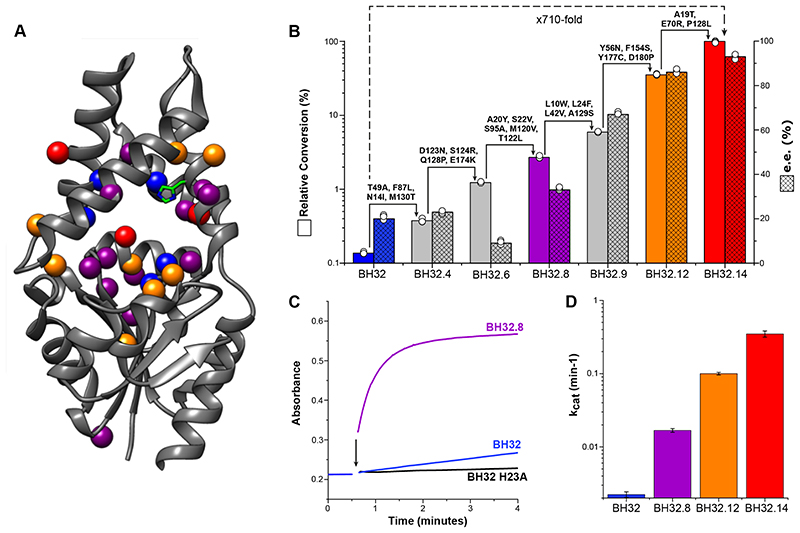
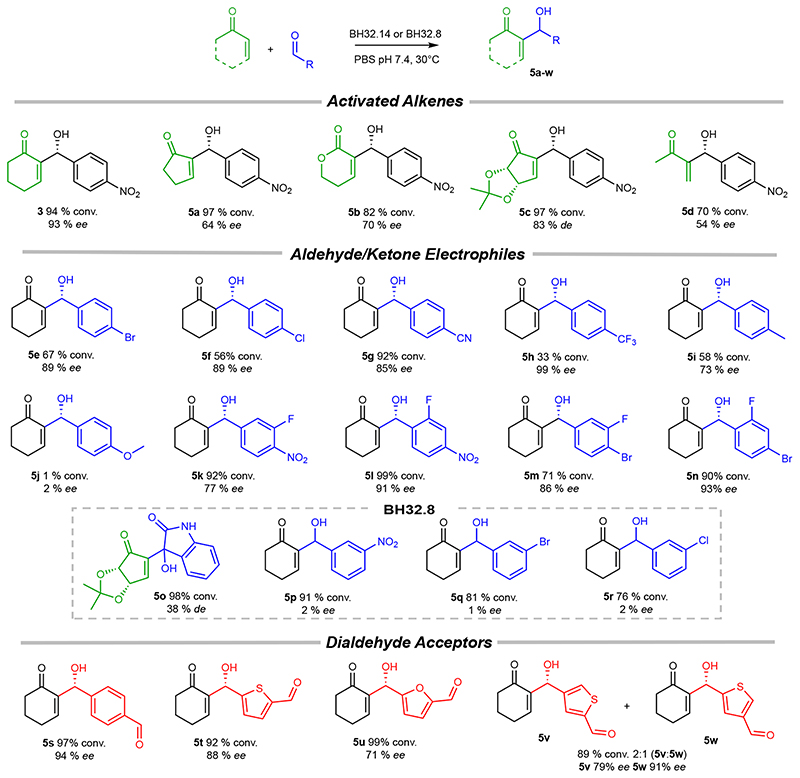
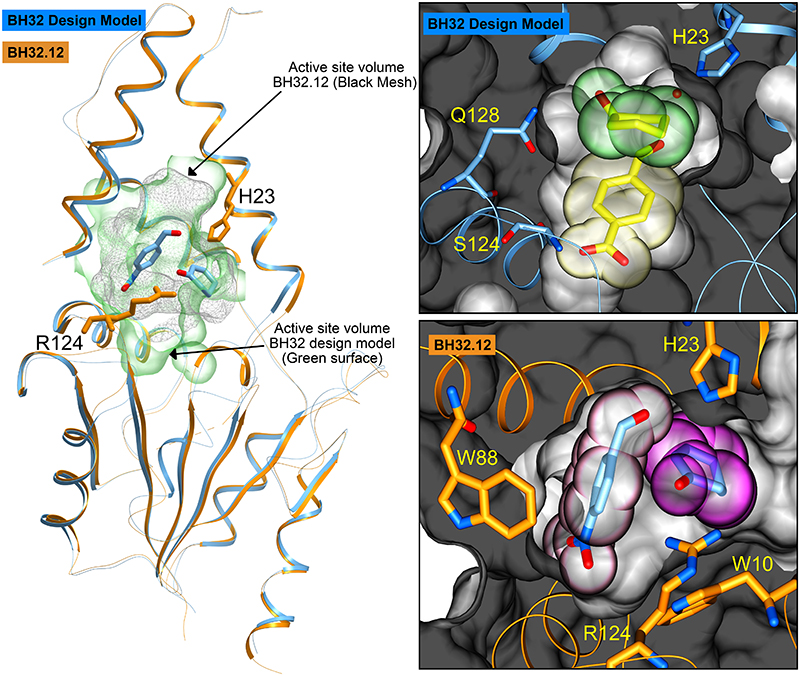
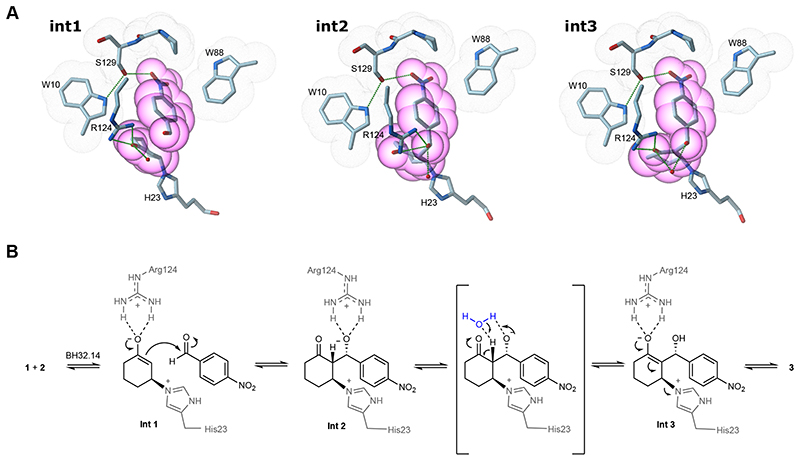
The combination of computational design and directed evolution could offer a general strategy to create enzymes with new functions. So far, this approach has delivered enzymes for a handful of model reactions. Here we show that new catalytic mechanisms can be engineered into proteins to accelerate more challenging chemical transformations. Evolutionary optimization of a primitive design afforded an efficient and enantioselective enzyme (BH32.14) for the Morita-Baylis-Hillman (MBH) reaction. BH32.14 is suitable for preparative-scale transformations, accepts a broad range of aldehyde and enone coupling partners and is able to promote selective monofunctionalizations of dialdehydes. Crystallographic, biochemical and computational studies reveal that BH32.14 operates via a sophisticated catalytic mechanism comprising a His23 nucleophile paired with a judiciously positioned Arg124. This catalytic arginine shuttles between conformational states to stabilize multiple oxyanion intermediates and serves as a genetically encoded surrogate of privileged bidentate hydrogen-bonding catalysts (for example, thioureas). This study demonstrates that elaborate catalytic devices can be built from scratch to promote demanding multi-step processes not observed in nature.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Building enzymes from scratch.Nat Chem. 2022 Mar;14(3):246-248. doi: 10.1038/s41557-021-00884-y. Nat Chem. 2022. PMID: 35102322 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- BB/M017702/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/S507040/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 757991/ERC_/European Research Council/International
- MR/T041722/1/MRC_/Medical Research Council/United Kingdom
- BB/M027023/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
