

Interleukin-2-inducible T-cell kinase (Itk) signaling regulates potent noncanonical regulatory T cells
- PMID: 34919342
- PMCID: PMC8679839
- DOI: 10.1002/ctm2.625
Interleukin-2-inducible T-cell kinase (Itk) signaling regulates potent noncanonical regulatory T cells
Abstract
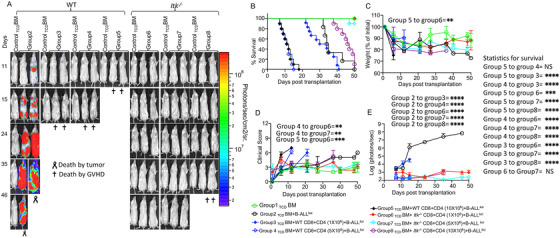
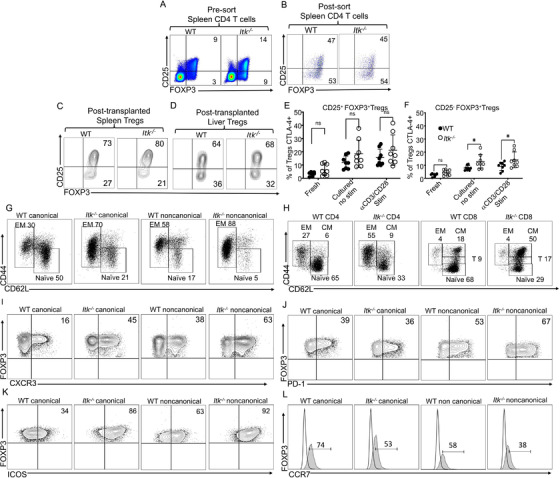
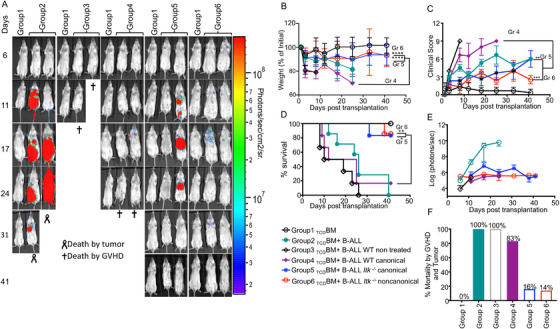
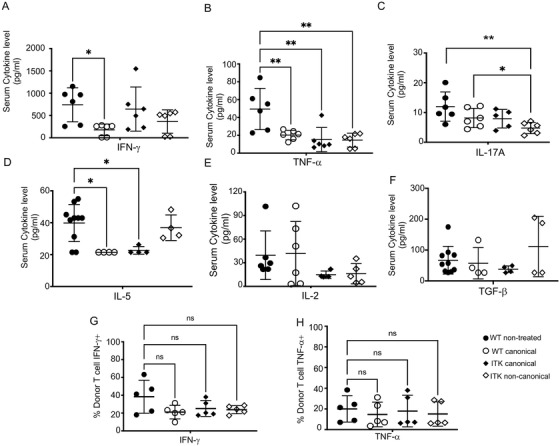
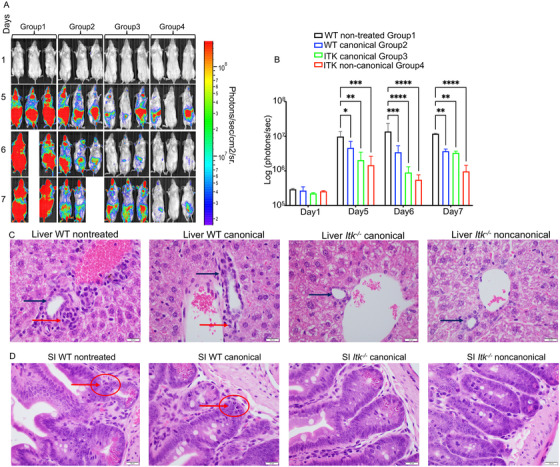
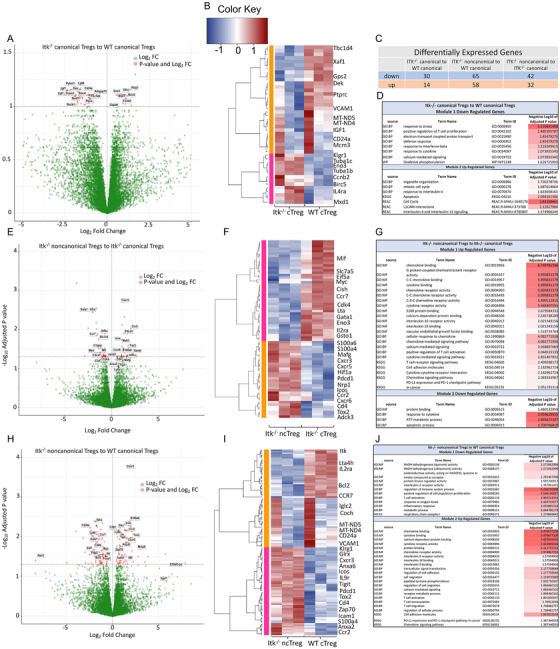
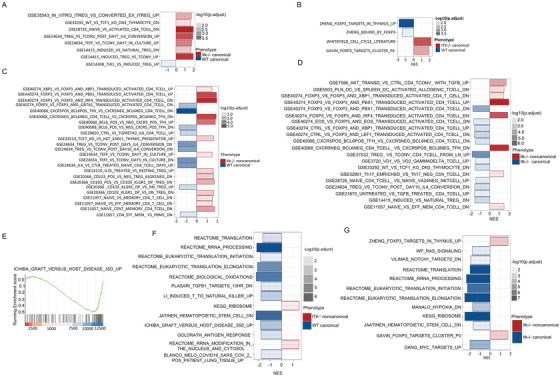
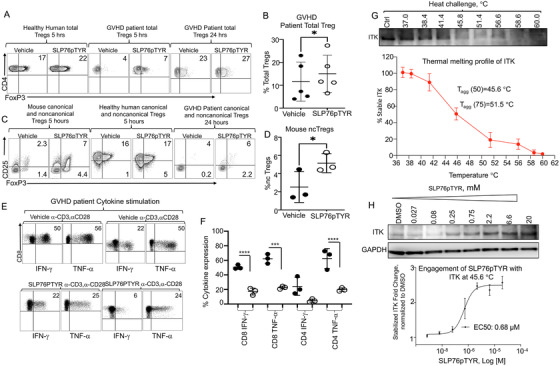
Regulatory T cells (Tregs) play an important role in controlling autoimmunity and limiting tissue damage and inflammation. IL2-inducible T cell kinase (Itk) is part of the Tec family of tyrosine kinases and is a critical component of T cell receptor mediated signaling. Here, we showed that either genetic ablation of Itk signaling or inhibition of Itk signaling pathways resulted in increased frequency of "noncanonical" CD4+ CD25- FOXP3+ Tregs (ncTregs), as well as of "canonical" CD4+ CD25+ FOXP3+ Tregs (canTregs). Using in vivo models, we showed that ncTregs can avert the formation of acute graft-versus-host disease (GVHD), in part by reducing conventional T cell proliferation, proinflammatory cytokine production, and tissue damage. This reduction in GVHD occurred without disruption of graft-versus-leukaemia (GVL) effects. RNA sequencing revealed that a number of effector, cell adhesion, and migration molecules were upregulated in Itk-/- ncTregs. Furthermore, disrupting the SLP76: ITK interaction using a specific peptide inhibitor led to enhanced Treg development in both mouse and primary human cells. This peptide inhibitor also significantly reduced inflammatory cytokine production in primary GVHD patient samples and mouse T cells without causing cell death or apoptosis. We provide evidence that specifically targeting Itk signaling could be a therapeutic strategy to treat autoimmune disorders.
Keywords: CCR7; CTLA-4; CXCR3; GVHD; GVL; ICOS; Itk; PD-1; SLP76pTYR; Tregs; canonical Tregs; noncanonical Tregs.
© 2021 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
AA receives research support from 3M Corporation. The other authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials