

A Summary of the Inaugural WHO Classification of Pediatric Tumors: Transitioning from the Optical into the Molecular Era
- PMID: 34921008
- PMCID: PMC9401511
- DOI: 10.1158/2159-8290.CD-21-1094
A Summary of the Inaugural WHO Classification of Pediatric Tumors: Transitioning from the Optical into the Molecular Era
Abstract
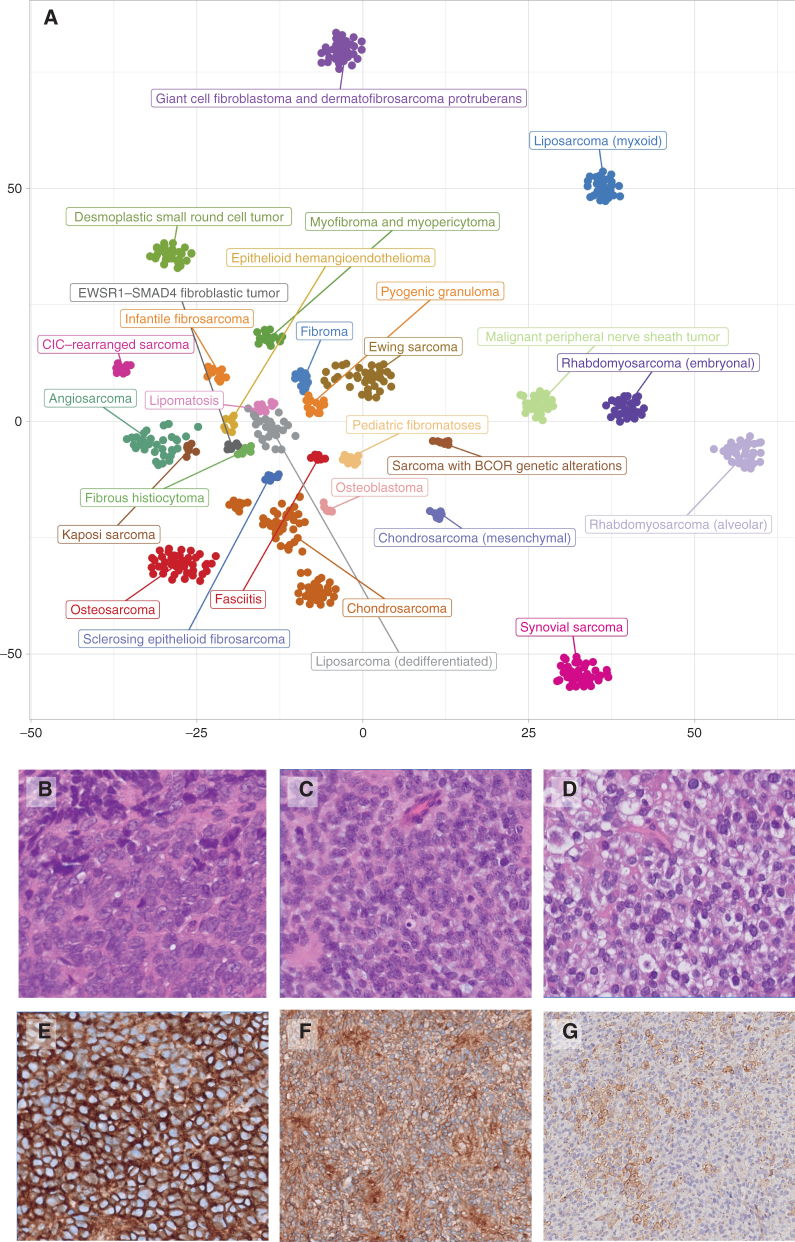
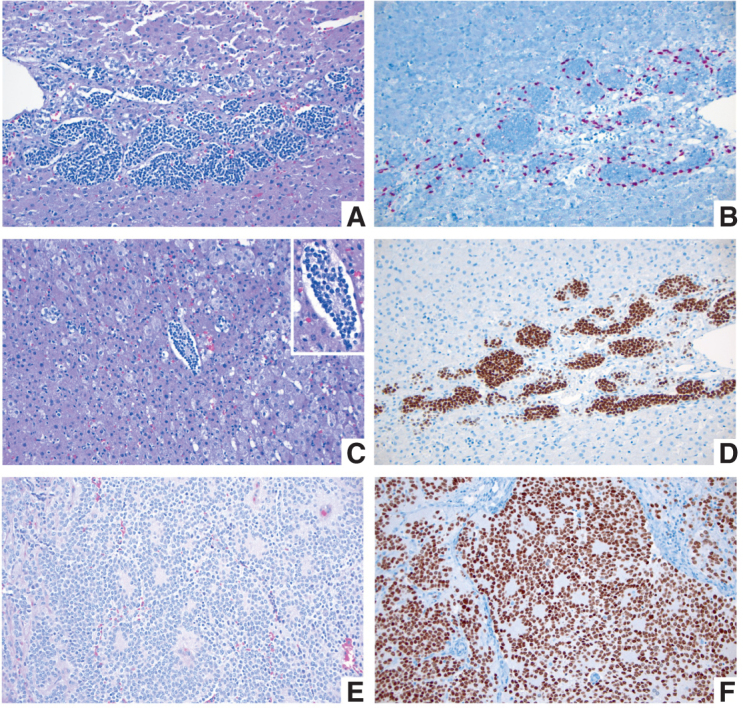
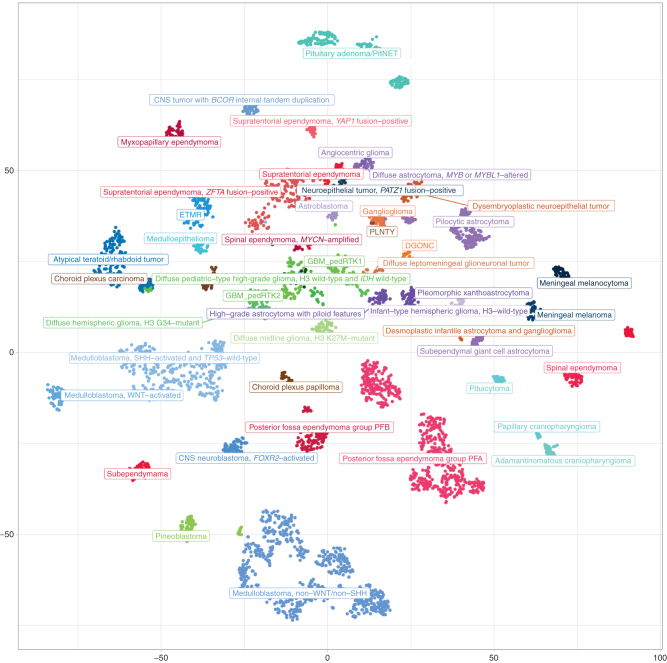
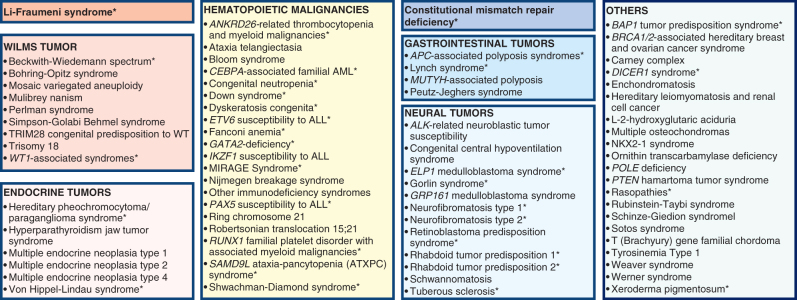
Pediatric tumors are uncommon, yet are the leading cause of cancer-related death in childhood. Tumor types, molecular characteristics, and pathogenesis are unique, often originating from a single genetic driver event. The specific diagnostic challenges of childhood tumors led to the development of the first World Health Organization (WHO) Classification of Pediatric Tumors. The classification is rooted in a multilayered approach, incorporating morphology, IHC, and molecular characteristics. The volume is organized according to organ sites and provides a single, state-of-the-art compendium of pediatric tumor types. A special emphasis was placed on "blastomas," which variably recapitulate the morphologic maturation of organs from which they originate. SIGNIFICANCE: In this review, we briefly summarize the main features and updates of each chapter of the inaugural WHO Classification of Pediatric Tumors, including its rapid transition from a mostly microscopic into a molecularly driven classification systematically taking recent discoveries in pediatric tumor genomics into account.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014;64:83–103. - PubMed
-
- Behjati S, Gilbertson RJ, Pfister SM. Maturation block in childhood cancer. Cancer Discov 2021;11:542–4. - PubMed
-
- Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VAet al. . The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
