

ddPCR allows 16S rRNA gene amplicon sequencing of very small DNA amounts from low-biomass samples
- PMID: 34922460
- PMCID: PMC8684222
- DOI: 10.1186/s12866-021-02391-z
ddPCR allows 16S rRNA gene amplicon sequencing of very small DNA amounts from low-biomass samples
Abstract
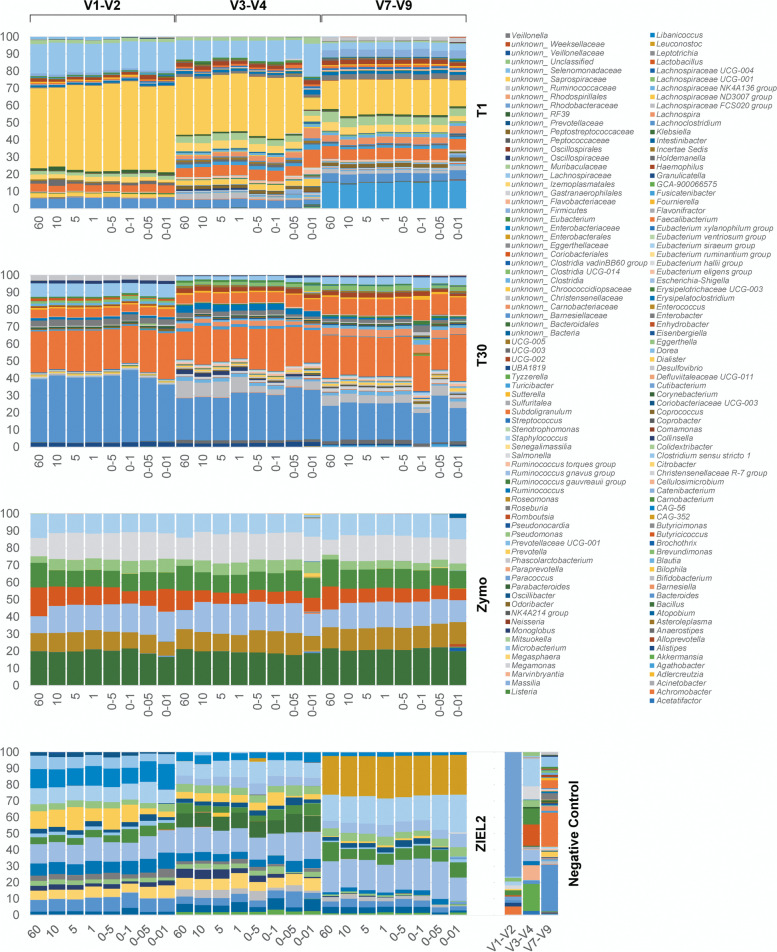
Background: One limiting factor of short amplicon 16S rRNA gene sequencing approaches is the use of low DNA amounts in the amplicon generation step. Especially for low-biomass samples, insufficient or even commonly undetectable DNA amounts can limit or prohibit further analysis in standard protocols.
Results: Using a newly established protocol, very low DNA input amounts were found sufficient for reliable detection of bacteria using 16S rRNA gene sequencing compared to standard protocols. The improved protocol includes an optimized amplification strategy by using a digital droplet PCR. We demonstrate how PCR products are generated even when using very low concentrated DNA, unable to be detected by using a Qubit. Importantly, the use of different 16S rRNA gene primers had a greater effect on the resulting taxonomical profiles compared to using high or very low initial DNA amounts.
Conclusion: Our improved protocol takes advantage of ddPCR and allows faithful amplification of very low amounts of template. With this, samples of low bacterial biomass become comparable to those with high amounts of bacteria, since the first and most biasing steps are the same. Besides, it is imperative to state DNA concentrations and volumes used and to include negative controls indicating possible shifts in taxonomical profiles. Despite this, results produced by using different primer pairs cannot be easily compared.
Keywords: 16S rRNA gene sequencing; Low-biomass samples; Very small DNA amounts; ddPCR.
© 2021. The Author(s).
Conflict of interest statement
KN has ongoing scientific collaborations with HiPP GmbH & Co. Vertrieb KG (Pfaffenhofen, Germany). All other authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
