

Neuropathology and molecular diagnosis of Synucleinopathies
- PMID: 34922583
- PMCID: PMC8684287
- DOI: 10.1186/s13024-021-00501-z
Neuropathology and molecular diagnosis of Synucleinopathies
Abstract
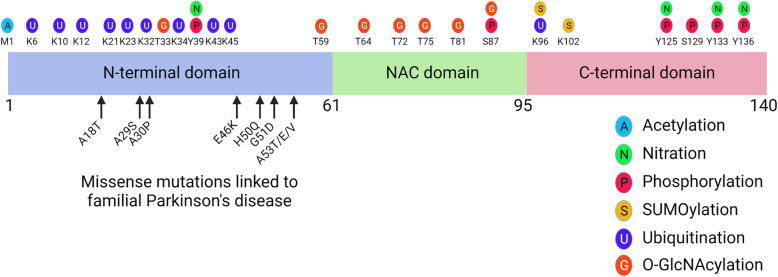
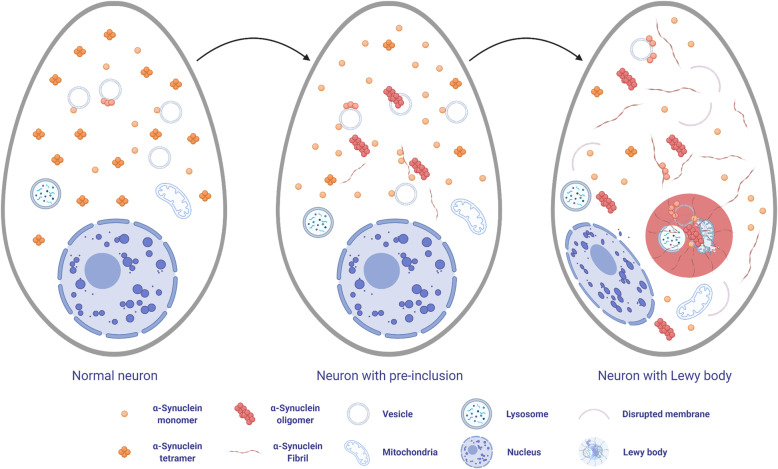
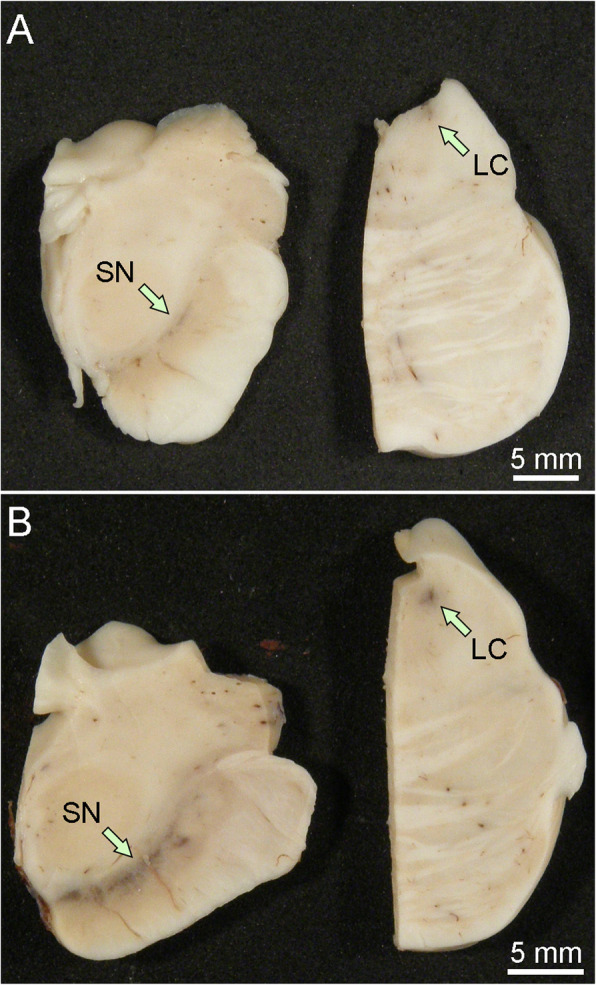
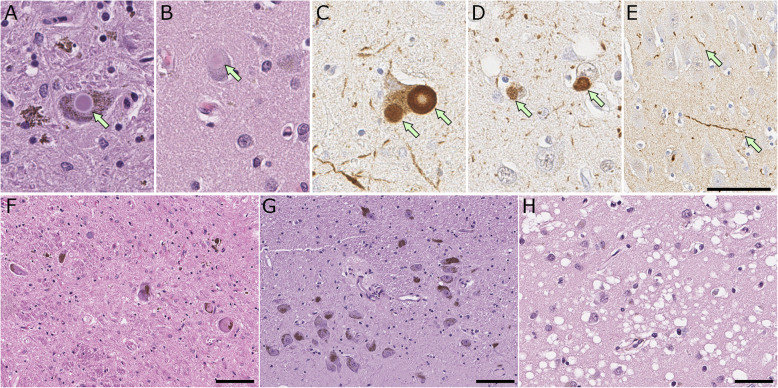
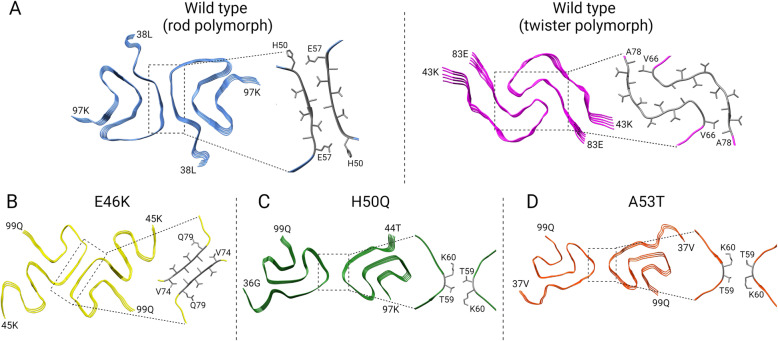
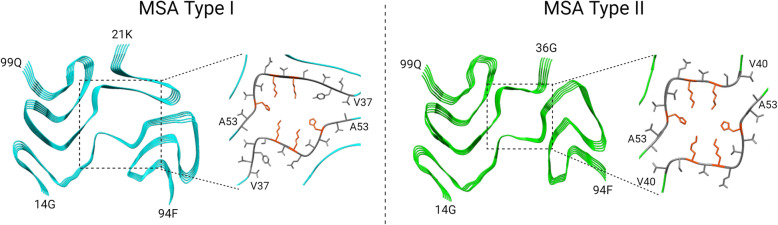
Synucleinopathies are clinically and pathologically heterogeneous disorders characterized by pathologic aggregates of α-synuclein in neurons and glia, in the form of Lewy bodies, Lewy neurites, neuronal cytoplasmic inclusions, and glial cytoplasmic inclusions. Synucleinopathies can be divided into two major disease entities: Lewy body disease and multiple system atrophy (MSA). Common clinical presentations of Lewy body disease are Parkinson's disease (PD), PD with dementia, and dementia with Lewy bodies (DLB), while MSA has two major clinical subtypes, MSA with predominant cerebellar ataxia and MSA with predominant parkinsonism. There are currently no disease-modifying therapies for the synucleinopathies, but information obtained from molecular genetics and models that explore mechanisms of α-synuclein conversion to pathologic oligomers and insoluble fibrils offer hope for eventual therapies. It remains unclear how α-synuclein can be associated with distinct cellular pathologies (e.g., Lewy bodies and glial cytoplasmic inclusions) and what factors determine neuroanatomical and cell type vulnerability. Accumulating evidence from in vitro and in vivo experiments suggests that α-synuclein species derived from Lewy body disease and MSA are distinct "strains" having different seeding properties. Recent advancements in in vitro seeding assays, such as real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA), not only demonstrate distinct seeding activity in the synucleinopathies, but also offer exciting opportunities for molecular diagnosis using readily accessible peripheral tissue samples. Cryogenic electron microscopy (cryo-EM) structural studies of α-synuclein derived from recombinant or brain-derived filaments provide new insight into mechanisms of seeding in synucleinopathies. In this review, we describe clinical, genetic and neuropathologic features of synucleinopathies, including a discussion of the evolution of classification and staging of Lewy body disease. We also provide a brief discussion on proposed mechanisms of Lewy body formation, as well as evidence supporting the existence of distinct α-synuclein strains in Lewy body disease and MSA.
Keywords: AlphaFold; Biomarkers; Dementia with Lewy bodies; Lewy body disease; Multiple system atrophy; PMCA; Parkinson’s disease; RT-QuIC; cryo-EM structures.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Hardy J, Gwinn-Hardy K. Genetic classification of primary neurodegenerative disease. Science. 1998;282:1075–1079. - PubMed
-
- Goedert M, Spillantini MG. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol Psychiatry. 1998;3:462–465. - PubMed
-
- Kosaka K, Matsushita M, Oyanagi S, Mehraein P. A clinico-neuropathological study of “Lewy body disease.” Psy-chiatry Neurol Jpn. 1980;82:292–311. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
