

Harmonizing variant classification for return of results in the All of Us Research Program
- PMID: 34923710
- PMCID: PMC9206690
- DOI: 10.1002/humu.24317
Harmonizing variant classification for return of results in the All of Us Research Program
Abstract
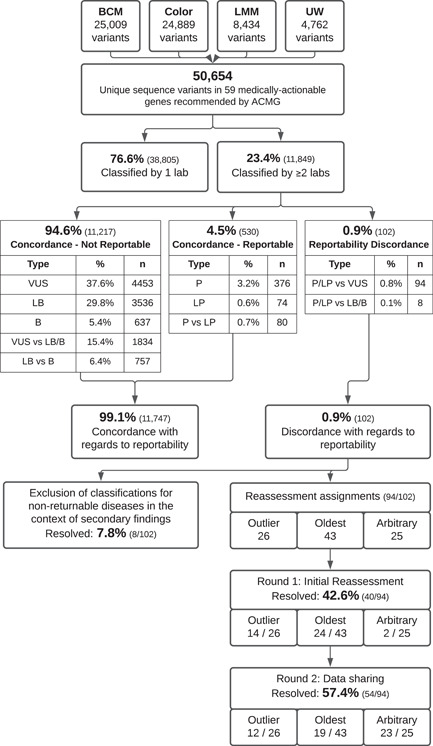
The All of Us Research Program (AoURP) is a historic effort to accelerate research and improve healthcare by generating and collating data from one million people in the United States. Participants will have the option to receive results from their genome analysis, including actionable findings in 59 gene-disorder pairs for which disorder-associated variants are recommended for return by the American College of Medical Genetics and Genomics. To ensure consistent reporting across the AoURP, in a prelaunch study the four participating clinical laboratories shared all variant classifications in the 59 genes of interest from their internal databases. Of the 11,813 unique variants classified by at least two of the four laboratories, classifications were concordant with regard to reportability for 99.1% (11,711), with only 0.9% (102) having reportability differences. Through variant reassessment, data sharing, and discussion of rationale, participating laboratories resolved all 102 reportable differences. These approaches will be maintained during routine AoU reporting to ensure continuous classification harmonization and consistent reporting within AoURP.
Keywords: All of Us Research Program; data sharing; variant classification.
© 2021 The Authors. Human Mutation published by Wiley Periodicals LLC.
Conflict of interest statement
The authors declare no additional conflicts of interest beyond their employment affiliation.
Figures
Similar articles
-
Scaling resolution of variant classification differences in ClinVar between 41 clinical laboratories through an outlier approach.Hum Mutat. 2018 Nov;39(11):1641-1649. doi: 10.1002/humu.23643. Hum Mutat. 2018. PMID: 30311378 Free PMC article.
-
Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar.Genet Med. 2017 Oct;19(10):1096-1104. doi: 10.1038/gim.2017.14. Epub 2017 Mar 16. Genet Med. 2017. PMID: 28301460 Free PMC article.
-
ClinGen's RASopathy Expert Panel consensus methods for variant interpretation.Genet Med. 2018 Nov;20(11):1334-1345. doi: 10.1038/gim.2018.3. Epub 2018 Mar 1. Genet Med. 2018. PMID: 29493581 Free PMC article.
-
The current state of clinical interpretation of sequence variants.Curr Opin Genet Dev. 2017 Feb;42:33-39. doi: 10.1016/j.gde.2017.01.001. Epub 2017 Jan 31. Curr Opin Genet Dev. 2017. PMID: 28157586 Free PMC article. Review.
-
Shariant platform: Enabling evidence sharing across Australian clinical genetic-testing laboratories to support variant interpretation.Am J Hum Genet. 2022 Nov 3;109(11):1960-1973. doi: 10.1016/j.ajhg.2022.10.006. Am J Hum Genet. 2022. PMID: 36332611 Free PMC article. Review.
Cited by
-
Estimating cancer risk in carriers of Lynch syndrome variants in UK Biobank.J Med Genet. 2024 Aug 29;61(9):861-869. doi: 10.1136/jmg-2023-109791. J Med Genet. 2024. PMID: 39004446 Free PMC article.
-
Recommendations for risk allele evidence curation, classification, and reporting from the ClinGen Low Penetrance/Risk Allele Working Group.Genet Med. 2024 Mar;26(3):101036. doi: 10.1016/j.gim.2023.101036. Epub 2023 Dec 3. Genet Med. 2024. PMID: 38054408 Free PMC article.
-
Genomic Analysis Identifies Risk Factors in Restless Legs Syndrome.Ann Neurol. 2024 Nov;96(5):994-1005. doi: 10.1002/ana.27040. Epub 2024 Jul 30. Ann Neurol. 2024. PMID: 39078117
-
Genomic analysis identifies risk factors in restless legs syndrome.medRxiv [Preprint]. 2023 Dec 20:2023.12.19.23300211. doi: 10.1101/2023.12.19.23300211. medRxiv. 2023. Update in: Ann Neurol. 2024 Nov;96(5):994-1005. doi: 10.1002/ana.27040. PMID: 38168192 Free PMC article. Updated. Preprint.
-
Using multiplexed functional data to reduce variant classification inequities in underrepresented populations.Genome Med. 2024 Dec 3;16(1):143. doi: 10.1186/s13073-024-01392-7. Genome Med. 2024. PMID: 39627863 Free PMC article.
References
-
- Amendola, L. M. , Jarvik, G. P. , Leo, M. C. , McLaughlin, H. M. , Akkari, Y. , Amaral, M. D. , Berg, J. S. , Biswas, S. , Bowling, K. M. , Conlin, L. K. , Cooper, G. M. , Dorschner, M. O. , Dulik, M. C. , Ghazani, A. A. , Ghosh, R. , Green, R. C. , Hart, R. , Horton, C. , Johnston, J. J. , & Rehm, H. L. (2016). Performance of ACMG‐AMP variant‐interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. American Journal of Human Genetics, 98(6), 1067–1076. 10.1016/j.ajhg.2016.03.024 - DOI - PMC - PubMed
-
- Amendola, L. M. , Muenzen, K. , Biesecker, L. G. , Bowling, K. M. , Cooper, G. M. , Dorschner, M. O. , Driscoll, C. , Foreman, A. K. M. , Golden‐Grant, K. , Greally, J. M. , Hindorff, L. , Kanavy, D. , Jobanputra, V. , Johnston, J. J. , Kenny, E. E. , McNulty, S. , Murali, P. , Ou, J. , Powell, B. C. , & Jarvik, G. P. (2020). Variant classification concordance using the ACMG‐AMP variant interpretation guidelines across nine genomic implementation research studies. American Journal of Human Genetics, 107(5), 932–941. 10.1016/j.ajhg.2020.09.011 - DOI - PMC - PubMed
-
- Chora, J. R. , Iacocca, M. A. , Tichy, L. , Wand, H. , Kurtz, C. L. , Zimmermann, H. , Leon, A. , Williams, M. , Humphries, S. E. , Hooper, A. J. , Trinder, M. , Brunham, L. R. , Pereira, A. C. , Jannes, C. E. , Chen, M. , Chonis, J. , Wang, J. , Kim, S. , Johnston, T. , & Bourbon, M. (2021). The clinical genome resource (ClinGen) familial hypercholesterolemia variant curation Expert Panel consensus guidelines for LDLR variant classification [Preprint]. Genetic and Genomic Medicine . 10.1101/2021.03.17.21252755 - DOI - PubMed
-
- Dewey, F. E. , Murray, M. F. , Overton, J. D. , Habegger, L. , Leader, J. B. , Fetterolf, S. N. , O'Dushlaine, C. , Van Hout, C. V. , Staples, J. , Gonzaga‐Jauregui, C. , Metpally, R. , Pendergrass, S. A. , Giovanni, M. A. , Kirchner, H. L. , Balasubramanian, S. , Abul‐Husn, N. S. , Hartzel, D. N. , Lavage, D. R. , Kost, K. A. , & Carey, D. J. (2016). Distribution and clinical impact of functional variants in 50,726 whole‐exome sequences from the DiscovEHR study. Science, 354(6319), aaf6814. 10.1126/science.aaf6814 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
