

: ComplexOme-Structural Network Interpreter used to study spatial enrichment in metazoan ribosomes
- PMID: 34930116
- PMCID: PMC8686616
- DOI: 10.1186/s12859-021-04510-z
: ComplexOme-Structural Network Interpreter used to study spatial enrichment in metazoan ribosomes
Abstract
Background: Upon environmental stimuli, ribosomes are surmised to undergo compositional rearrangements due to abundance changes among proteins assembled into the complex, leading to modulated structural and functional characteristics. Here, we present the ComplexOme-Structural Network Interpreter (), a computational method to allow testing whether ribosomal proteins (rProteins) that exhibit abundance changes under specific conditions are spatially confined to particular regions within the large ribosomal complex.
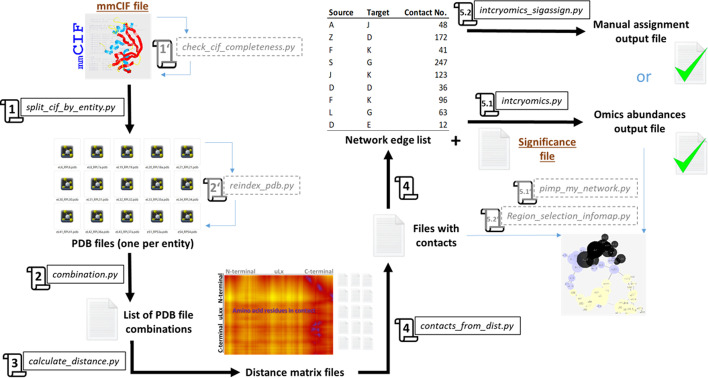
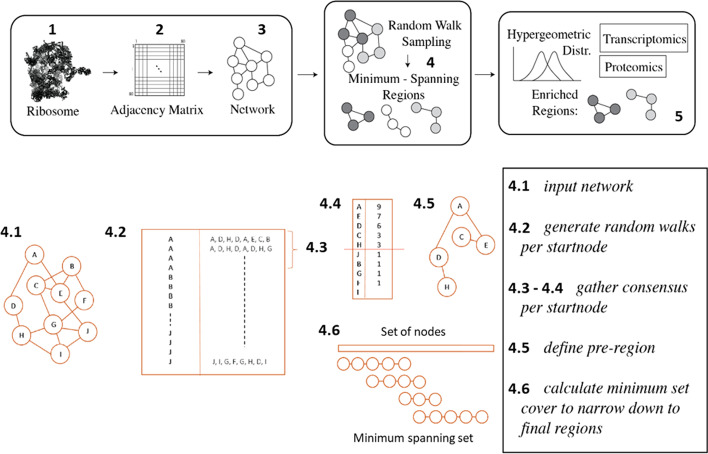
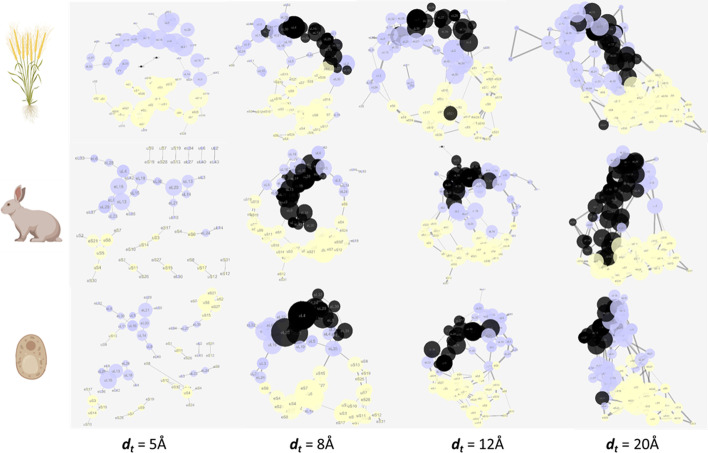
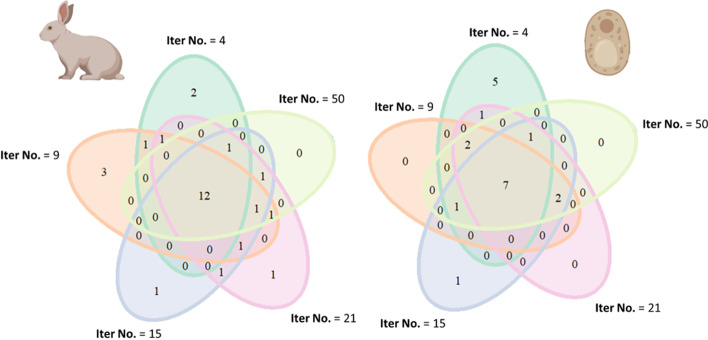
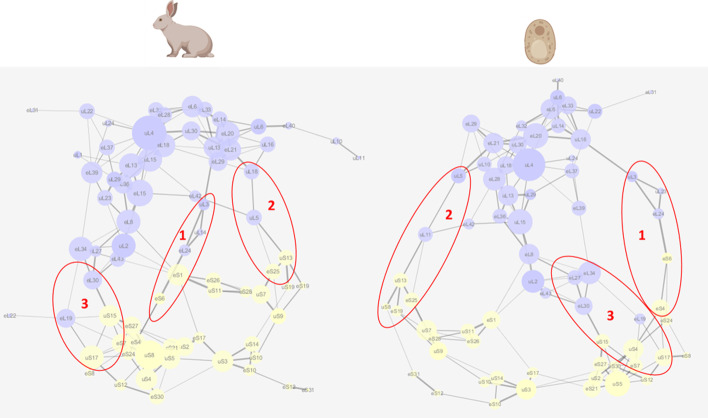
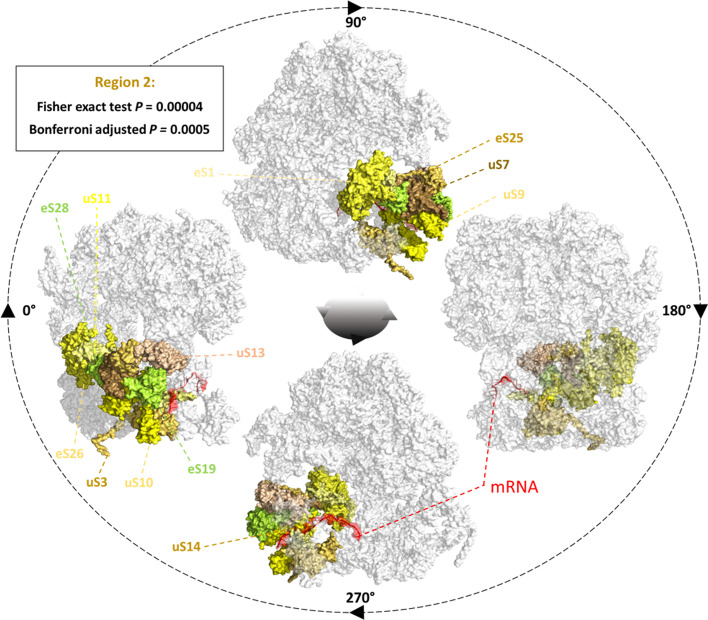
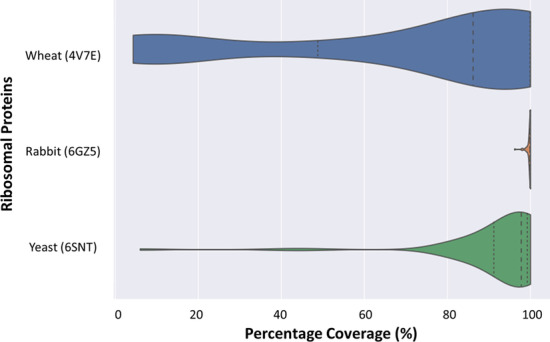
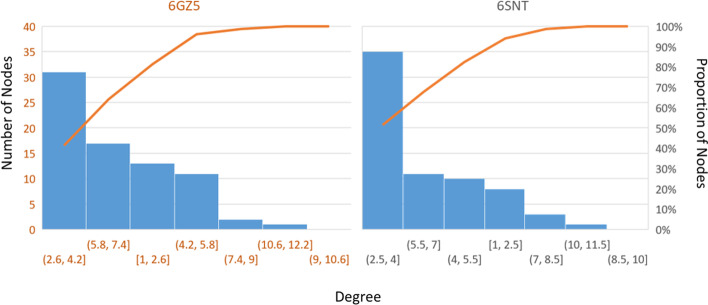
Results: translates experimentally determined structures into graphs, with nodes representing proteins and edges the spatial proximity between them. In its first implementation, considers rProteins and ignores rRNA and other objects. Spatial regions are defined using a random walk with restart methodology, followed by a procedure to obtain a minimum set of regions that cover all proteins in the complex. Structural coherence is achieved by applying weights to the edges reflecting the physical proximity between purportedly contacting proteins. The weighting probabilistically guides the random-walk path trajectory. Parameter tuning during region selection provides the option to tailor the method to specific biological questions by yielding regions of different sizes with minimum overlaps. In addition, other graph community detection algorithms may be used for the workflow, considering that they yield different sized, non-overlapping regions. All tested algorithms result in the same node kernels under equivalent regions. Based on the defined regions, available abundance change information of proteins is mapped onto the graph and subsequently tested for enrichment in any of the defined spatial regions. We applied to the cytosolic ribosome structures of Saccharomyces cerevisiae, Oryctolagus cuniculus, and Triticum aestivum using datasets with available quantitative protein abundance change information. We found that in yeast, substoichiometric rProteins depleted from translating polysomes are significantly constrained to a ribosomal region close to the tRNA entry and exit sites.
Conclusions: offers a computational method to partition multi-protein complexes into structural regions and a statistical approach to test for spatial enrichments of any given subsets of proteins. is applicable to any multi-protein complex given appropriate structural and abundance-change data. is publicly available as a GitHub repository https://github.com/MSeidelFed/COSNet_i and can be installed using the python installer pip.
Keywords: Omics integration; Ribosomal protein substoichiometry; Ribosome structure; Specialized ribosomes; Structural systems biology.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Streamlining Protein Fractional Synthesis Rates Using SP3 Beads and Stable Isotope Mass Spectrometry: A Case Study on the Plant Ribosome.Bio Protoc. 2024 May 5;14(9):e4981. doi: 10.21769/BioProtoc.4981. eCollection 2024 May 5. Bio Protoc. 2024. PMID: 38737506 Free PMC article.
-
Spatially Enriched Paralog Rearrangements Argue Functionally Diverse Ribosomes Arise during Cold Acclimation in Arabidopsis.Int J Mol Sci. 2021 Jun 7;22(11):6160. doi: 10.3390/ijms22116160. Int J Mol Sci. 2021. PMID: 34200446 Free PMC article.
-
Zero-Shot Video Object Segmentation With Co-Attention Siamese Networks.IEEE Trans Pattern Anal Mach Intell. 2022 Apr;44(4):2228-2242. doi: 10.1109/TPAMI.2020.3040258. Epub 2022 Mar 4. IEEE Trans Pattern Anal Mach Intell. 2022. PMID: 33232224
-
Ribosome biogenesis in the yeast Saccharomyces cerevisiae.Genetics. 2013 Nov;195(3):643-81. doi: 10.1534/genetics.113.153197. Genetics. 2013. PMID: 24190922 Free PMC article. Review.
-
Three-dimensional electron cryomicroscopy of ribosomes.Curr Protein Pept Sci. 2002 Feb;3(1):79-91. doi: 10.2174/1389203023380873. Curr Protein Pept Sci. 2002. PMID: 12370013 Review.
Cited by
-
Dynamics of ribosome composition and ribosomal protein phosphorylation in immune signaling in Arabidopsis thaliana.Nucleic Acids Res. 2023 Nov 27;51(21):11876-11892. doi: 10.1093/nar/gkad827. Nucleic Acids Res. 2023. PMID: 37823590 Free PMC article.
-
Remodelled ribosomal populations synthesize a specific proteome in proliferating plant tissue during cold.Philos Trans R Soc Lond B Biol Sci. 2025 Mar 6;380(1921):20230384. doi: 10.1098/rstb.2023.0384. Epub 2025 Mar 6. Philos Trans R Soc Lond B Biol Sci. 2025. PMID: 40045790 Free PMC article.
-
Streamlining Protein Fractional Synthesis Rates Using SP3 Beads and Stable Isotope Mass Spectrometry: A Case Study on the Plant Ribosome.Bio Protoc. 2024 May 5;14(9):e4981. doi: 10.21769/BioProtoc.4981. eCollection 2024 May 5. Bio Protoc. 2024. PMID: 38737506 Free PMC article.
References
-
- Baßler J, Hurt E. Eukaryotic ribosome assembly. Annu Rev Biochem. 2019;88:281–306. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
