Mitochondrial C5aR1 activity in macrophages controls IL-1β production underlying sterile inflammation
- PMID: 34932384
- PMCID: PMC8902698
- DOI: 10.1126/sciimmunol.abf2489
Mitochondrial C5aR1 activity in macrophages controls IL-1β production underlying sterile inflammation
Abstract
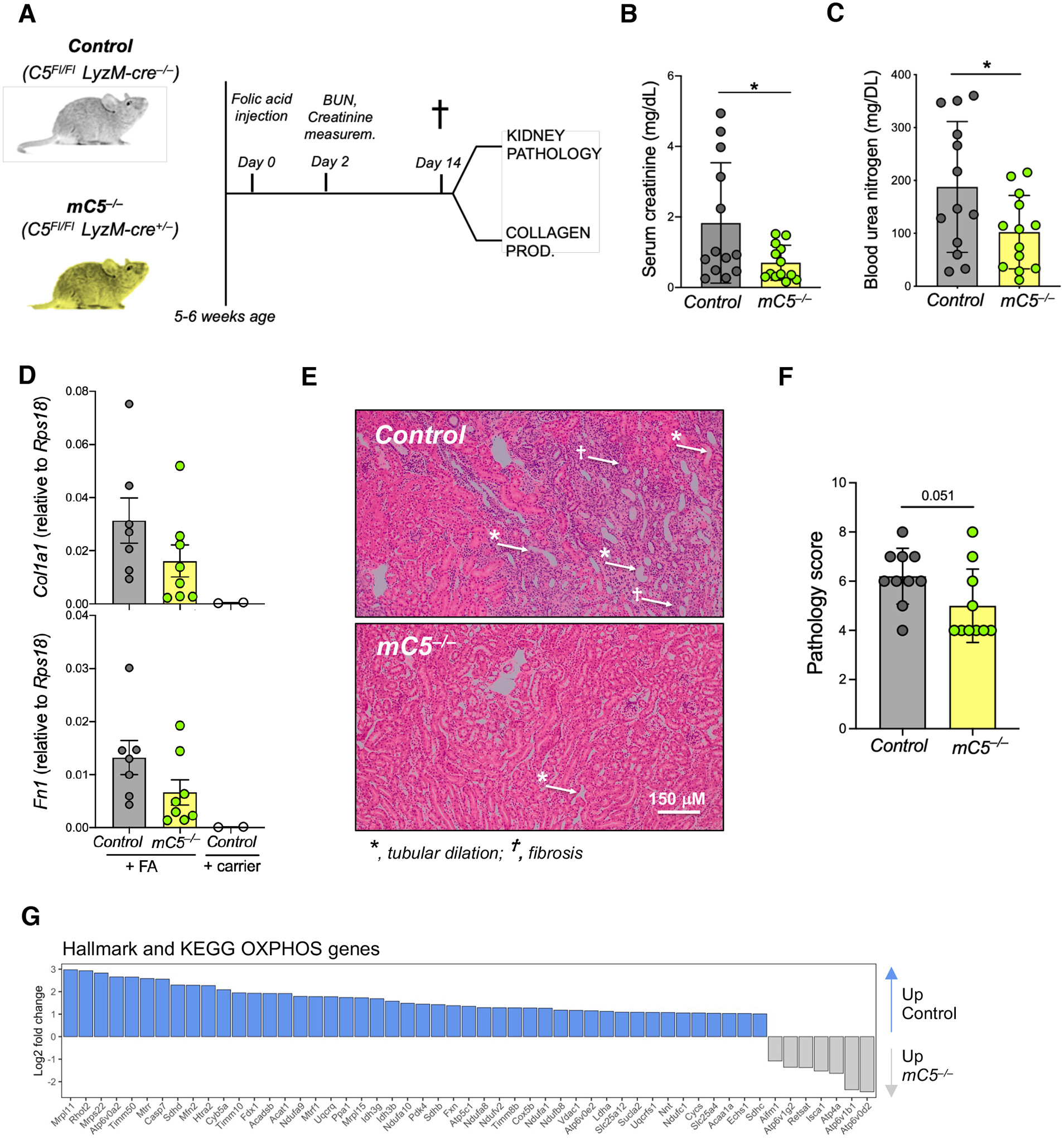
While serum-circulating complement destroys invading pathogens, intracellularly active complement, termed the “complosome,” functions as a vital orchestrator of cell-metabolic events underlying T cell effector responses. Whether intracellular complement is also nonredundant for the activity of myeloid immune cells is currently unknown. Here, we show that monocytes and macrophages constitutively express complement component (C) 5 and generate autocrine C5a via formation of an intracellular C5 convertase. Cholesterol crystal sensing by macrophages induced C5aR1 signaling on mitochondrial membranes, which shifted ATP production via reverse electron chain flux toward reactive oxygen species generation and anaerobic glycolysis to favor IL-1β production, both at the transcriptional level and processing of pro–IL-1β. Consequently, atherosclerosis-prone mice lacking macrophage-specific C5ar1 had ameliorated cardiovascular disease on a high-cholesterol diet. Conversely, inflammatory gene signatures and IL-1β produced by cells in unstable atherosclerotic plaques of patients were normalized by a specific cell-permeable C5aR1 antagonist. Deficiency of the macrophage cell-autonomous C5 system also protected mice from crystal nephropathy mediated by folic acid. These data demonstrate the unexpected intracellular formation of a C5 convertase and identify C5aR1 as a direct modulator of mitochondrial function and inflammatory output from myeloid cells. Together, these findings suggest that the complosome is a contributor to the biologic processes underlying sterile inflammation and indicate that targeting this system could be beneficial in macrophage-dependent diseases, such as atherosclerosis.
Conflict of interest statement
Figures







References
-
- Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, Arstila TP, Pekkarinen PT, Ma M, Cope A, Reinheckel T, Rodriguez de Cordoba S, Afzali B, Atkinson JP, Kemper C, Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157 (2013). - PMC - PubMed
-
- Arbore G, West EE, Rahman J, Le Friec G, Niyonzima N, Pirooznia M, Tunc I, Pavlidis P, Powell N, Li Y, Liu P, Servais A, Couzi L, Fremeaux-Bacchi V, Placais L, Ferraro A, Walsh PR, Kavanagh D, Afzali B, Lavender P, Lachmann HJ, Kemper C, Complement receptor CD46 co-stimulates optimal human CD8(+) T cell effector function via fatty acid metabolism. Nat.Commun 9, 4186 (2018). - PMC - PubMed
-
- Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, Fischer M, Belle R, Loeliger J, Develioglu L, Bantug GR, Watson J, Couzi L, Afzali B, Lavender P, Hess C, Kemper C, Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 42, 1033–1047 (2015). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous