

Activation mechanism of PINK1
- PMID: 34933320
- PMCID: PMC8828467
- DOI: 10.1038/s41586-021-04340-2
Activation mechanism of PINK1
Erratum in
-
Publisher Correction: Activation mechanism of PINK1.Nature. 2022 Mar;603(7903):E33. doi: 10.1038/s41586-022-04591-7. Nature. 2022. PMID: 35293391 Free PMC article. No abstract available.
Abstract
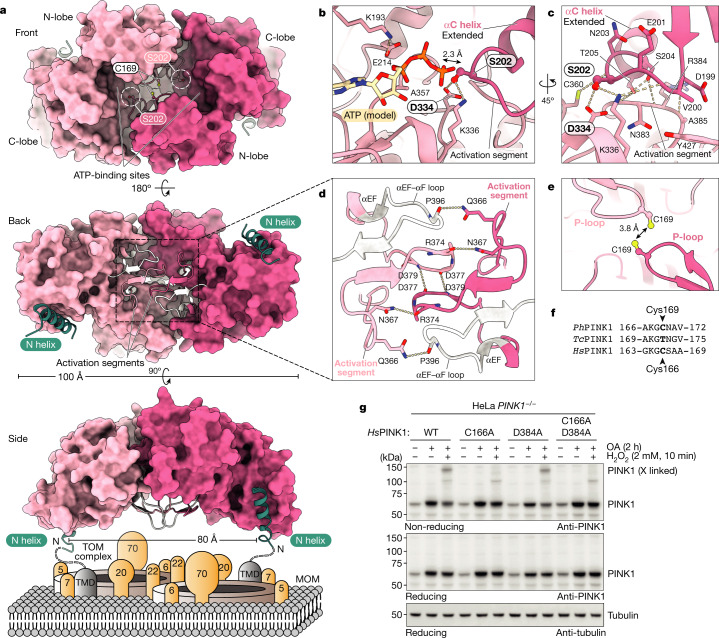
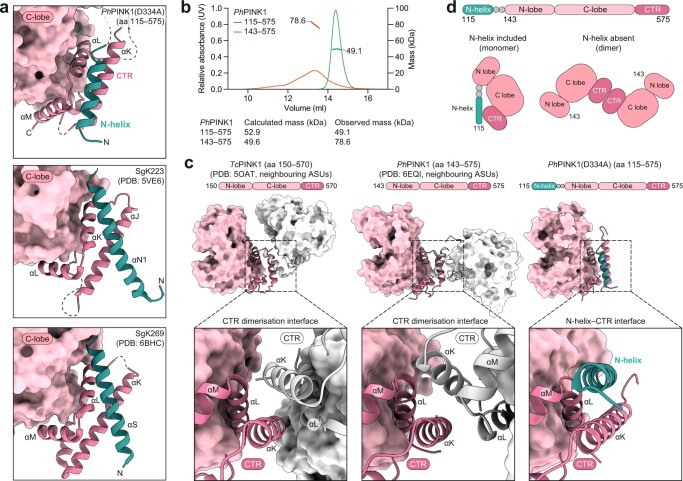
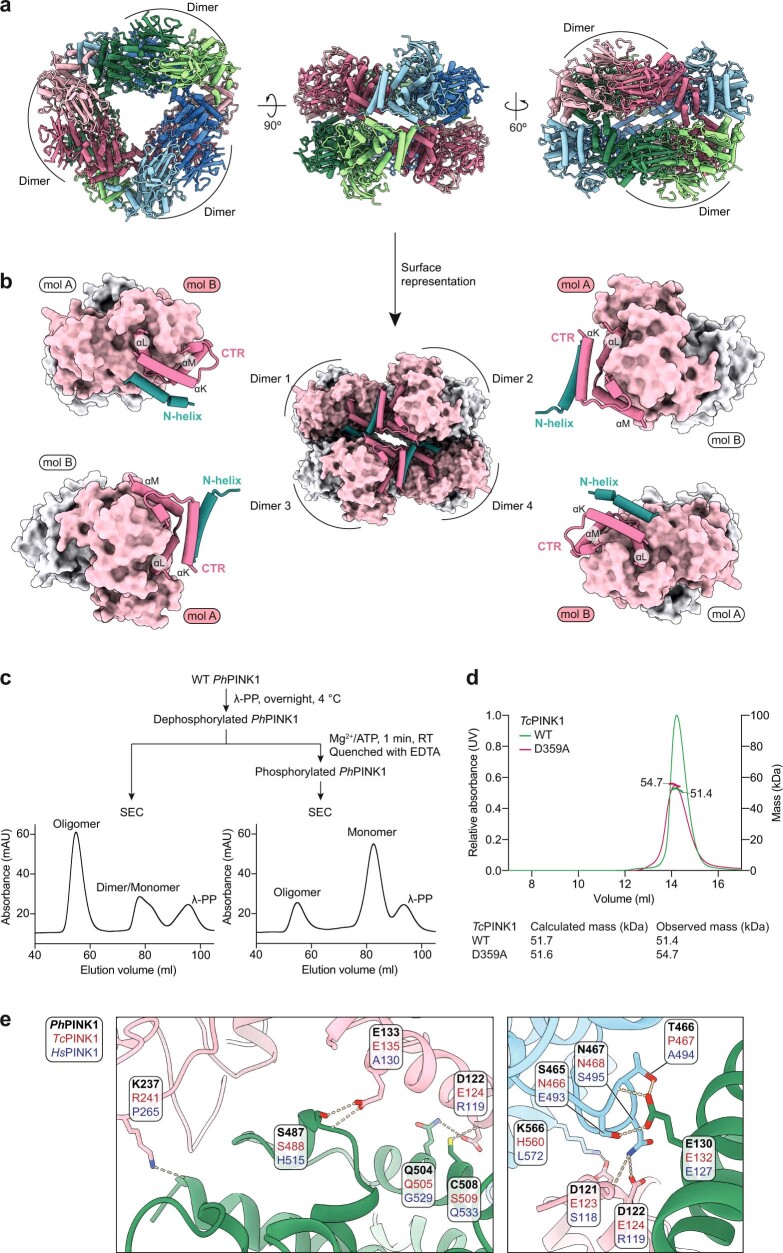
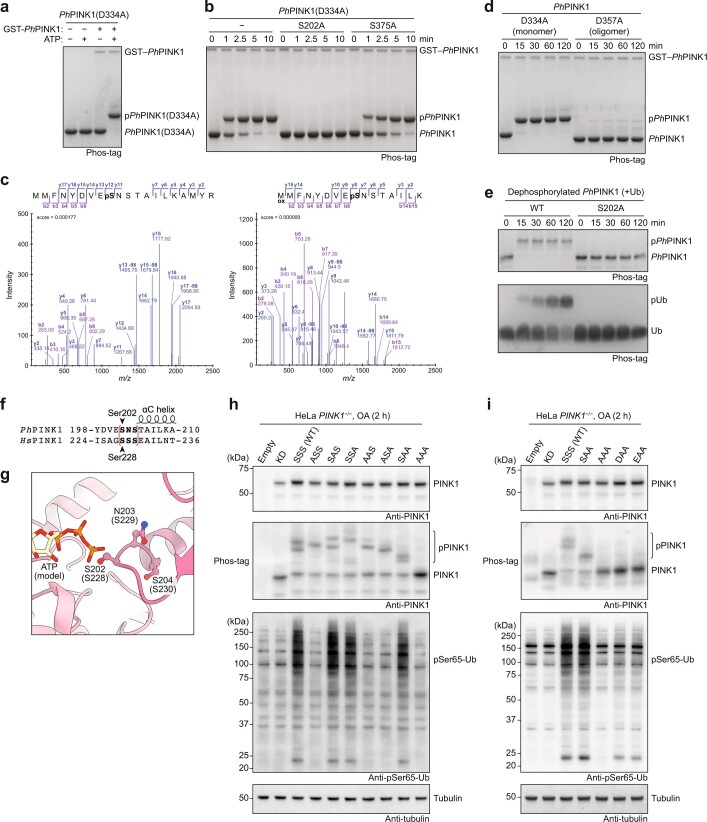
Mutations in the protein kinase PINK1 lead to defects in mitophagy and cause autosomal recessive early onset Parkinson's disease1,2. PINK1 has many unique features that enable it to phosphorylate ubiquitin and the ubiquitin-like domain of Parkin3-9. Structural analysis of PINK1 from diverse insect species10-12 with and without ubiquitin provided snapshots of distinct structural states yet did not explain how PINK1 is activated. Here we elucidate the activation mechanism of PINK1 using crystallography and cryo-electron microscopy (cryo-EM). A crystal structure of unphosphorylated Pediculus humanus corporis (Ph; human body louse) PINK1 resolves an N-terminal helix, revealing the orientation of unphosphorylated yet active PINK1 on the mitochondria. We further provide a cryo-EM structure of a symmetric PhPINK1 dimer trapped during the process of trans-autophosphorylation, as well as a cryo-EM structure of phosphorylated PhPINK1 undergoing a conformational change to an active ubiquitin kinase state. Structures and phosphorylation studies further identify a role for regulatory PINK1 oxidation. Together, our research delineates the complete activation mechanism of PINK1, illuminates how PINK1 interacts with the mitochondrial outer membrane and reveals how PINK1 activity may be modulated by mitochondrial reactive oxygen species.
© 2021. The Author(s).
Conflict of interest statement
D.K. serves on the Scientific Advisory Board of BioTheryX Inc. The other authors declare no competing interests.
Figures










References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
