

Genotype and Cardiac Outcomes in Pediatric Dilated Cardiomyopathy
- PMID: 34935411
- PMCID: PMC9075202
- DOI: 10.1161/JAHA.121.022854
Genotype and Cardiac Outcomes in Pediatric Dilated Cardiomyopathy
Abstract
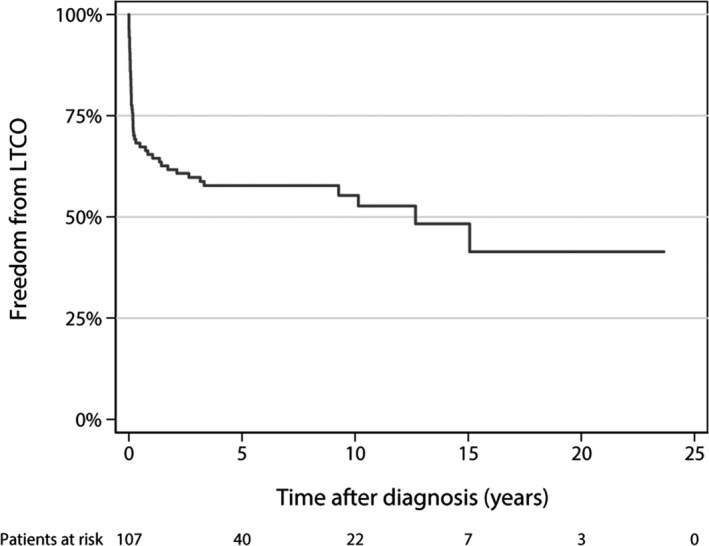
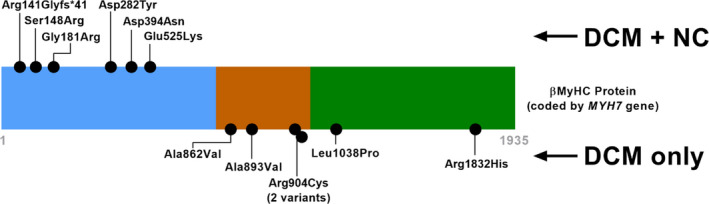
Background Pediatric dilated cardiomyopathy (DCM) is a well-known clinical entity; however, phenotype-genotype correlations are inadequately described. Our objective was to provide genotype associations with life-threatening cardiac outcomes in pediatric DCM probands. Methods and Results We performed a retrospective review of children with DCM at a large pediatric referral center (2007-2016), excluding syndromic, chemotherapy-induced, and congenital heart disease causes. Genetic variants were adjudicated by an expert panel and an independent clinical laboratory. In a cohort of 109 pediatric DCM cases with a mean age at diagnosis of 4.2 years (SD 5.9), life-threatening cardiac outcomes occurred in 47% (42% heart transplant, 5% death). One or more pathogenic/likely pathogenic variants were present in 40/109 (37%), and 36/44 (82%) of pathogenic/likely pathogenic variants occurred in sarcomeric genes. The frequency of pathogenic/likely pathogenic variants was not different in patients with familial cardiomyopathy (15/33 with family history versus 25/76 with no family history, P=0.21). TTN truncating variants occurred in a higher percentage of children diagnosed as teenagers (26% teenagers versus 6% younger children, P=0.01), but life-threatening cardiac outcomes occurred in both infants and teenagers with these TTN variants. DCM with left ventricular noncompaction features occurred in 6/6 patients with MYH7 variants between amino acids 1 and 600. Conclusions Sarcomeric variants were common in pediatric DCM. We demonstrated genotype-specific associations with age of diagnosis and cardiac outcomes. In particular, MYH7 had domain-specific association with DCM with left ventricular noncompaction features. Family history did not predict pathogenic/likely pathogenic variants, reinforcing that genetic testing should be considered in all children with idiopathic DCM.
Keywords: dilated cardiomyopathy; genotype; pediatrics; transplant.
Figures

References
-
- Rossano JW, Dipchand AI, Edwards LB, Goldfarb S, Kucheryavaya AY, Levvey BJ, Lung LH, Meiser B, Yusen RD, Stehlik J. The registry of the International Society for Heart and Lung Transplantation: nineteenth pediatric heart transplantation report‐2016; focus theme: primary diagnostic indications for transplant. J Heart Lung Transplant. 2016;35:1185–1195. doi: 10.1016/j.healun.2016.08.018 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
