

TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion
- PMID: 34935874
- PMCID: PMC8704919
- DOI: 10.1084/jem.20201966
TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion
Abstract
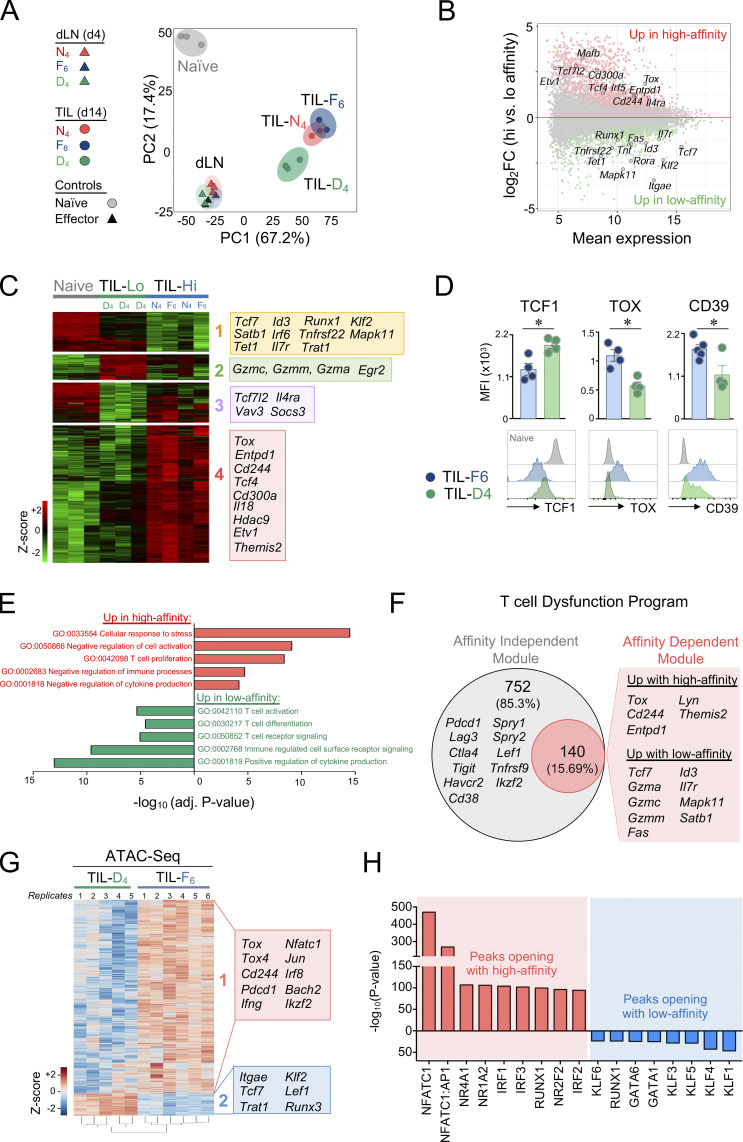
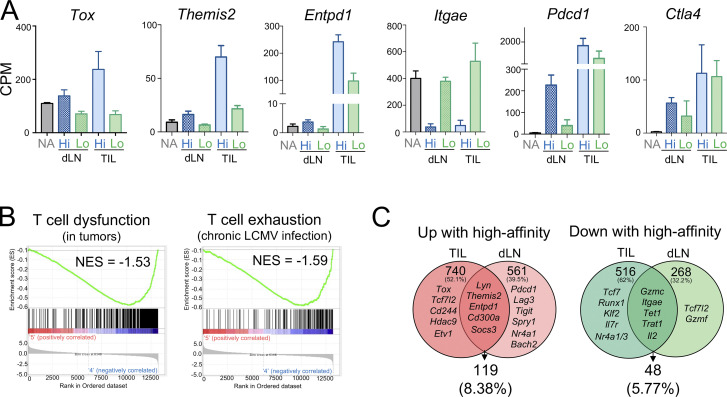
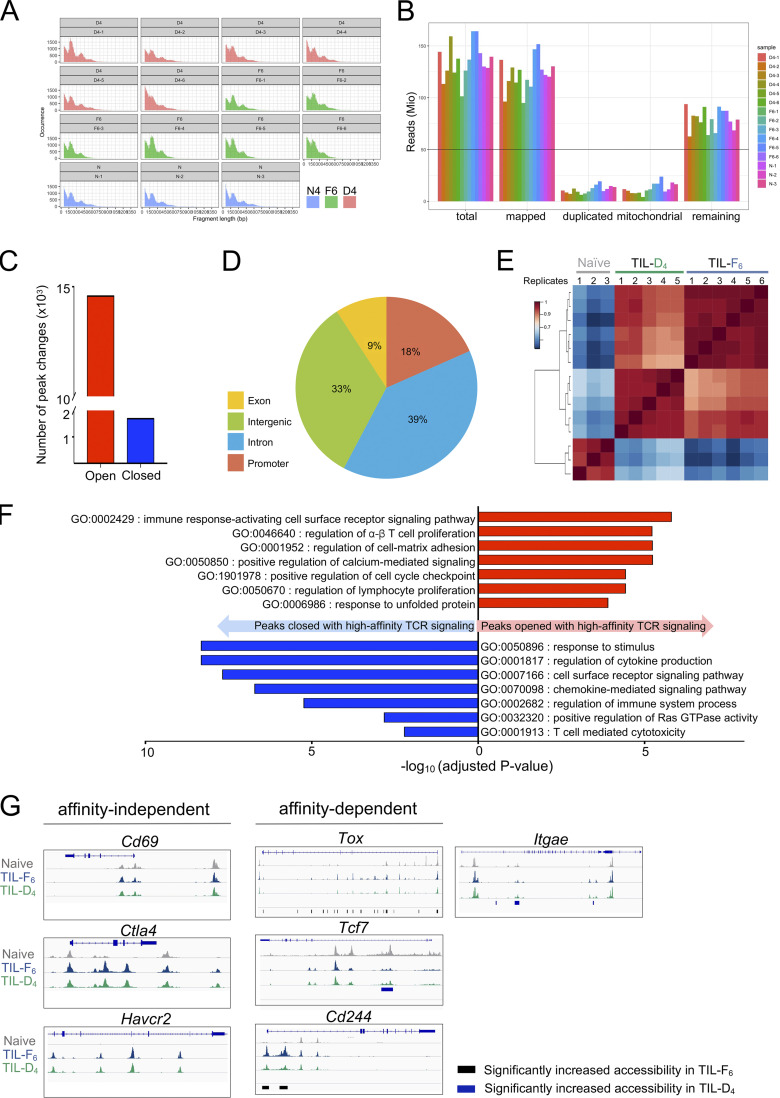
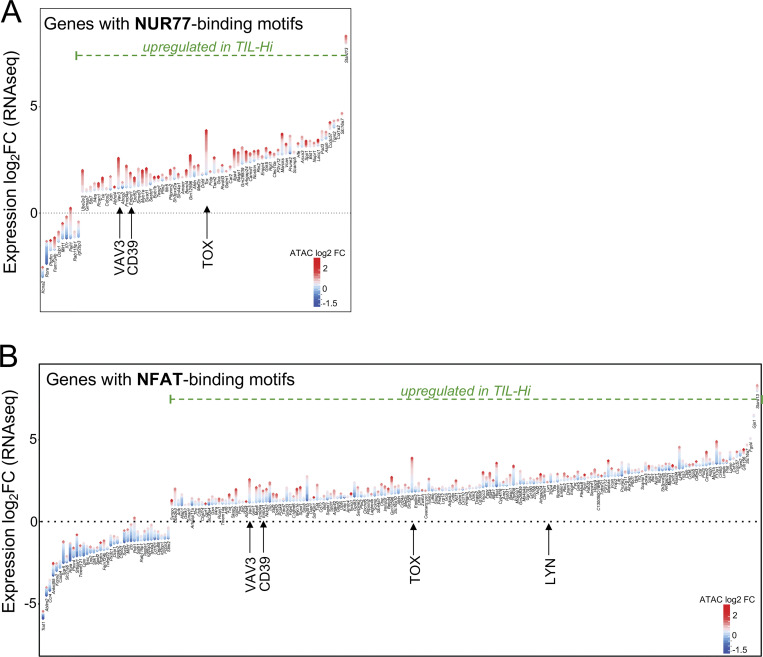
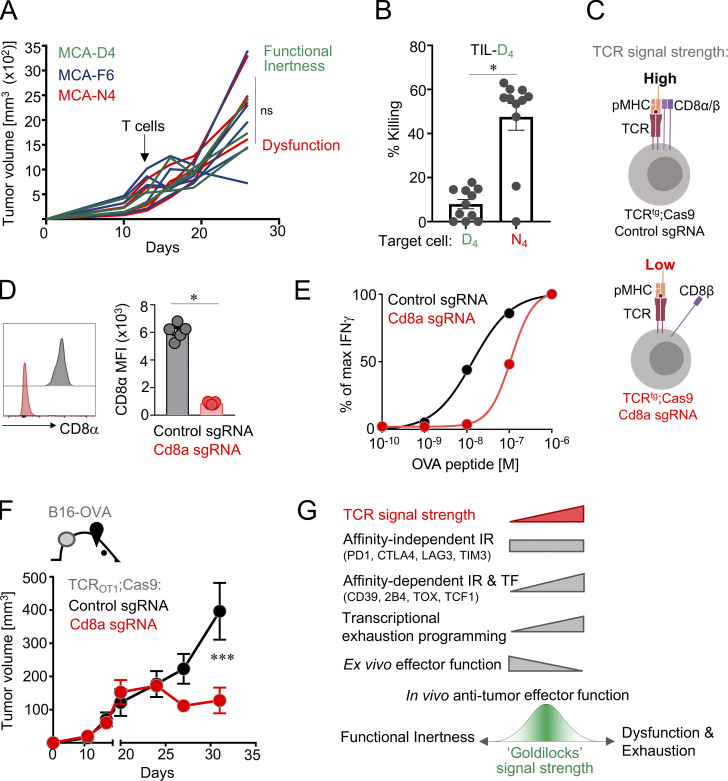
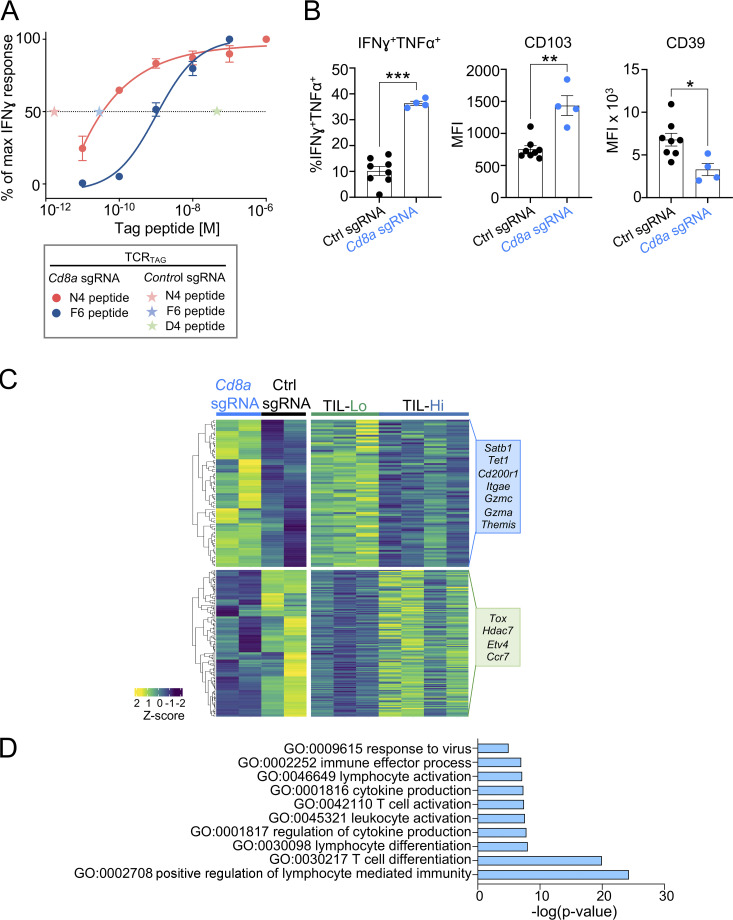
T cell receptor (TCR) signal strength is a key determinant of T cell responses. We developed a cancer mouse model in which tumor-specific CD8 T cells (TST cells) encounter tumor antigens with varying TCR signal strength. High-signal-strength interactions caused TST cells to up-regulate inhibitory receptors (IRs), lose effector function, and establish a dysfunction-associated molecular program. TST cells undergoing low-signal-strength interactions also up-regulated IRs, including PD1, but retained a cell-intrinsic functional state. Surprisingly, neither high- nor low-signal-strength interactions led to tumor control in vivo, revealing two distinct mechanisms by which PD1hi TST cells permit tumor escape; high signal strength drives dysfunction, while low signal strength results in functional inertness, where the signal strength is too low to mediate effective cancer cell killing by functional TST cells. CRISPR-Cas9-mediated fine-tuning of signal strength to an intermediate range improved anti-tumor activity in vivo. Our study defines the role of TCR signal strength in TST cell function, with important implications for T cell-based cancer immunotherapies.
© 2021 Shakiba et al.
Conflict of interest statement
Disclosures: N.A. Defranoux reports personal fees from Alector outside the submitted work. M.D. Hellman receives institutional research funding from Bristol-Myers Squibb; has been a compensated consultant for Merck, Bristol-Myers Squibb, AstraZeneca, Genentech/Roche, Nektar, Syndax, Mirati, Shattuck Labs, Immunai, Blueprint Medicines, Achilles, and Arcus; received travel support/honoraria from AstraZeneca, Eli Lilly, and Bristol-Myers Squibb; has options from Factorial, Shattuck Labs, Immunai, and Arcus; reported personal fees from Adicet, DaVolterra, Genzyme/Sanofi, Janssen, Immunai, Instil Bio, Mana Therapeutics, Natera, Pact Pharms, Chattuck Labs, and Regenron out side the submitted work; and has a patent filed by his institution related to the use of tumor mutation burden to predict response to immunotherapy (PCT/US2015/062208), which has received licensing fees from PGDx. D.K. Wells is a founder, equity holder, and receives advisory fees from Immunai. No other disclosures were reported.
Figures
Comment in
-
Goldilocks and the three TILs.J Exp Med. 2022 Feb 7;219(2):e20212269. doi: 10.1084/jem.20212269. Epub 2022 Jan 12. J Exp Med. 2022. PMID: 35020792 Free PMC article.
References
-
- Andrews, S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
