

MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra
- PMID: 34944436
- PMCID: PMC8699281
- DOI: 10.3390/biom11121793
MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra
Abstract
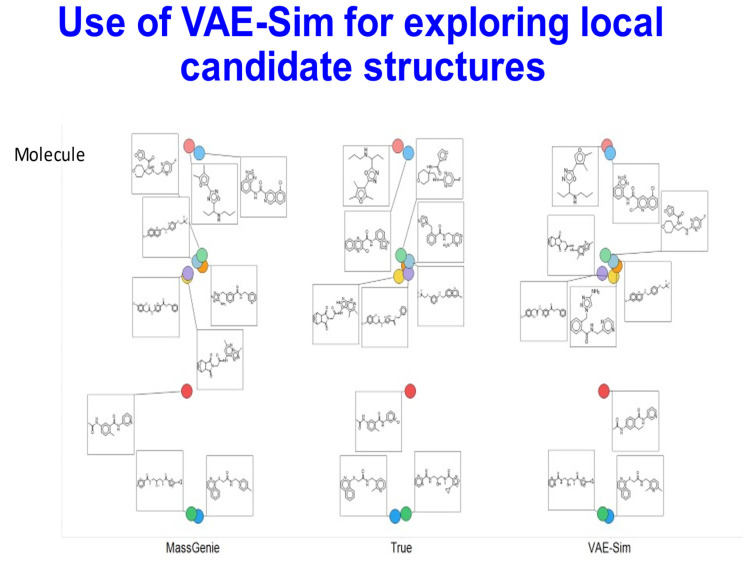
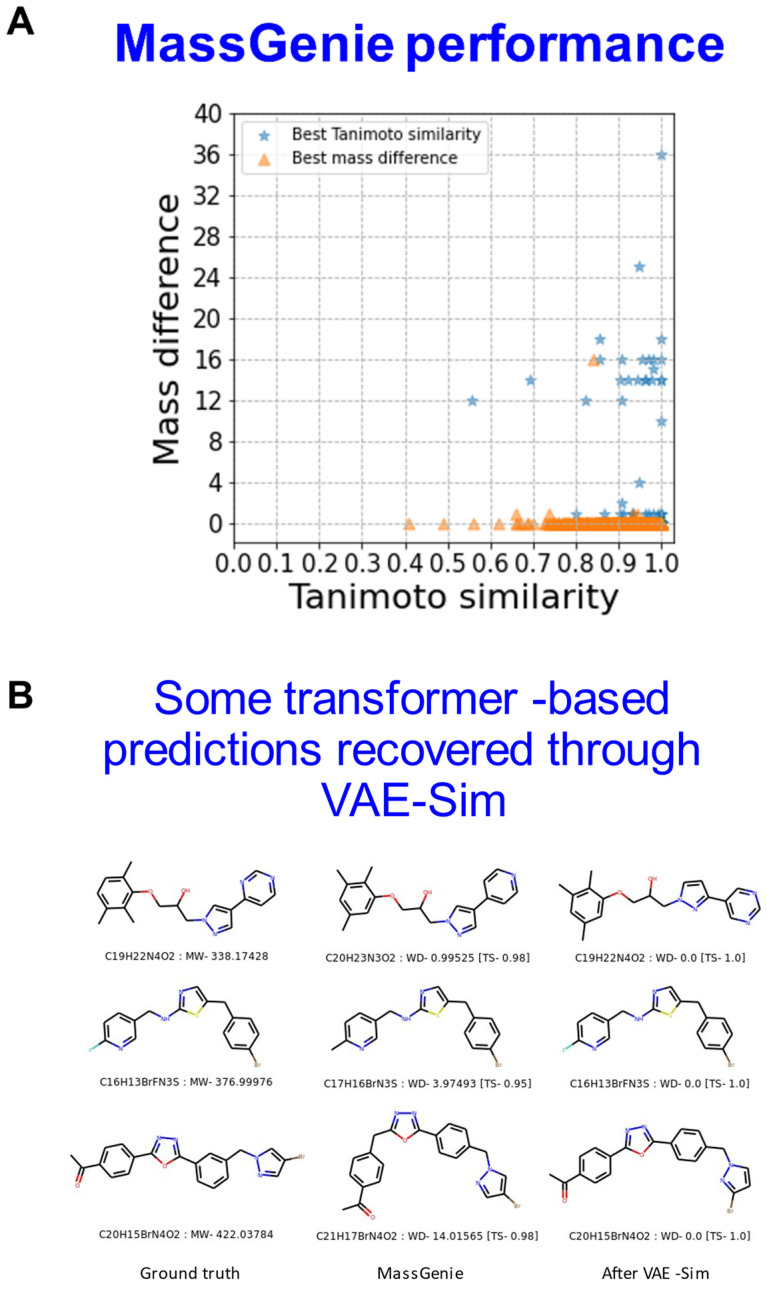
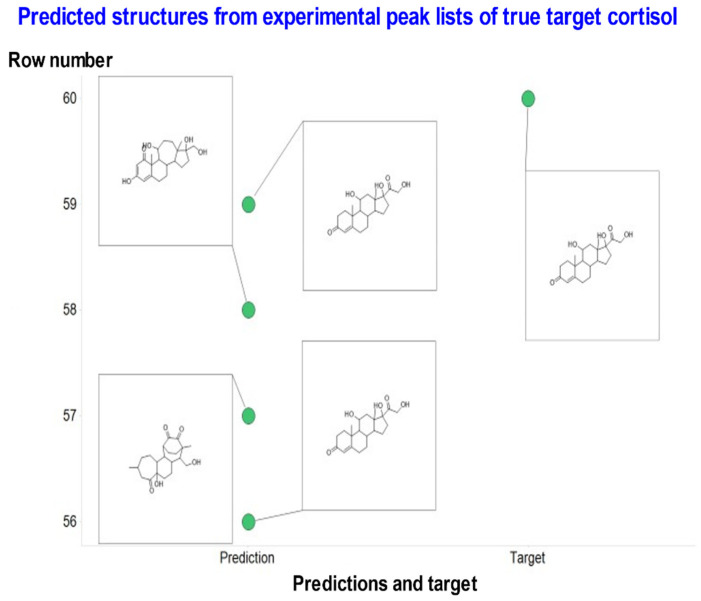
The 'inverse problem' of mass spectrometric molecular identification ('given a mass spectrum, calculate/predict the 2D structure of the molecule whence it came') is largely unsolved, and is especially acute in metabolomics where many small molecules remain unidentified. This is largely because the number of experimentally available electrospray mass spectra of small molecules is quite limited. However, the forward problem ('calculate a small molecule's likely fragmentation and hence at least some of its mass spectrum from its structure alone') is much more tractable, because the strengths of different chemical bonds are roughly known. This kind of molecular identification problem may be cast as a language translation problem in which the source language is a list of high-resolution mass spectral peaks and the 'translation' a representation (for instance in SMILES) of the molecule. It is thus suitable for attack using the deep neural networks known as transformers. We here present MassGenie, a method that uses a transformer-based deep neural network, trained on ~6 million chemical structures with augmented SMILES encoding and their paired molecular fragments as generated in silico, explicitly including the protonated molecular ion. This architecture (containing some 400 million elements) is used to predict the structure of a molecule from the various fragments that may be expected to be observed when some of its bonds are broken. Despite being given essentially no detailed nor explicit rules about molecular fragmentation methods, isotope patterns, rearrangements, neutral losses, and the like, MassGenie learns the effective properties of the mass spectral fragment and valency space, and can generate candidate molecular structures that are very close or identical to those of the 'true' molecules. We also use VAE-Sim, a previously published variational autoencoder, to generate candidate molecules that are 'similar' to the top hit. In addition to using the 'top hits' directly, we can produce a rank order of these by 'round-tripping' candidate molecules and comparing them with the true molecules, where known. As a proof of principle, we confine ourselves to positive electrospray mass spectra from molecules with a molecular mass of 500Da or lower, including those in the last CASMI challenge (for which the results are known), getting 49/93 (53%) precisely correct. The transformer method, applied here for the first time to mass spectral interpretation, works extremely effectively both for mass spectra generated in silico and on experimentally obtained mass spectra from pure compounds. It seems to act as a Las Vegas algorithm, in that it either gives the correct answer or simply states that it cannot find one. The ability to create and to 'learn' millions of fragmentation patterns in silico, and therefrom generate candidate structures (that do not have to be in existing libraries) directly, thus opens up entirely the field of de novo small molecule structure prediction from experimental mass spectra.
Keywords: artificial intelligence; chemical space; deep learning; electrospray; generative methods; mass spectrometry; metabolomics; transformers.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
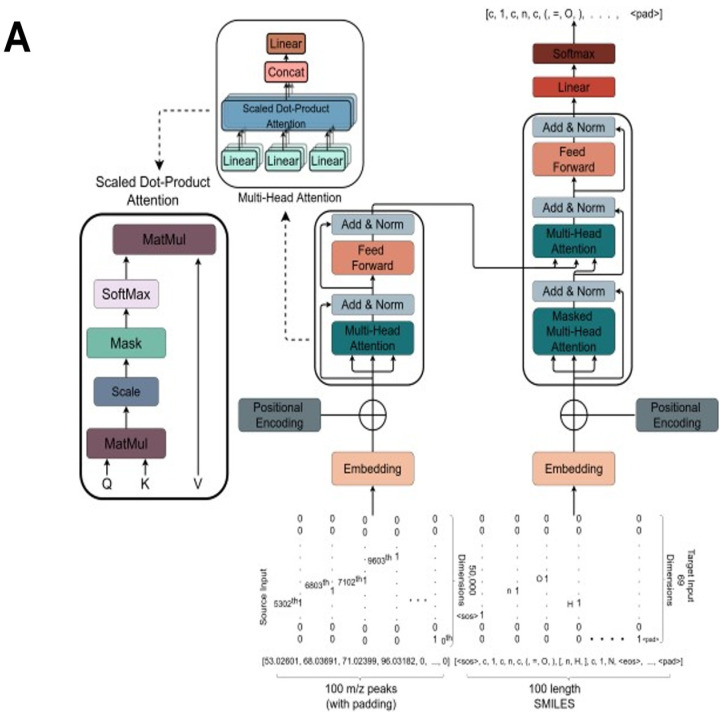
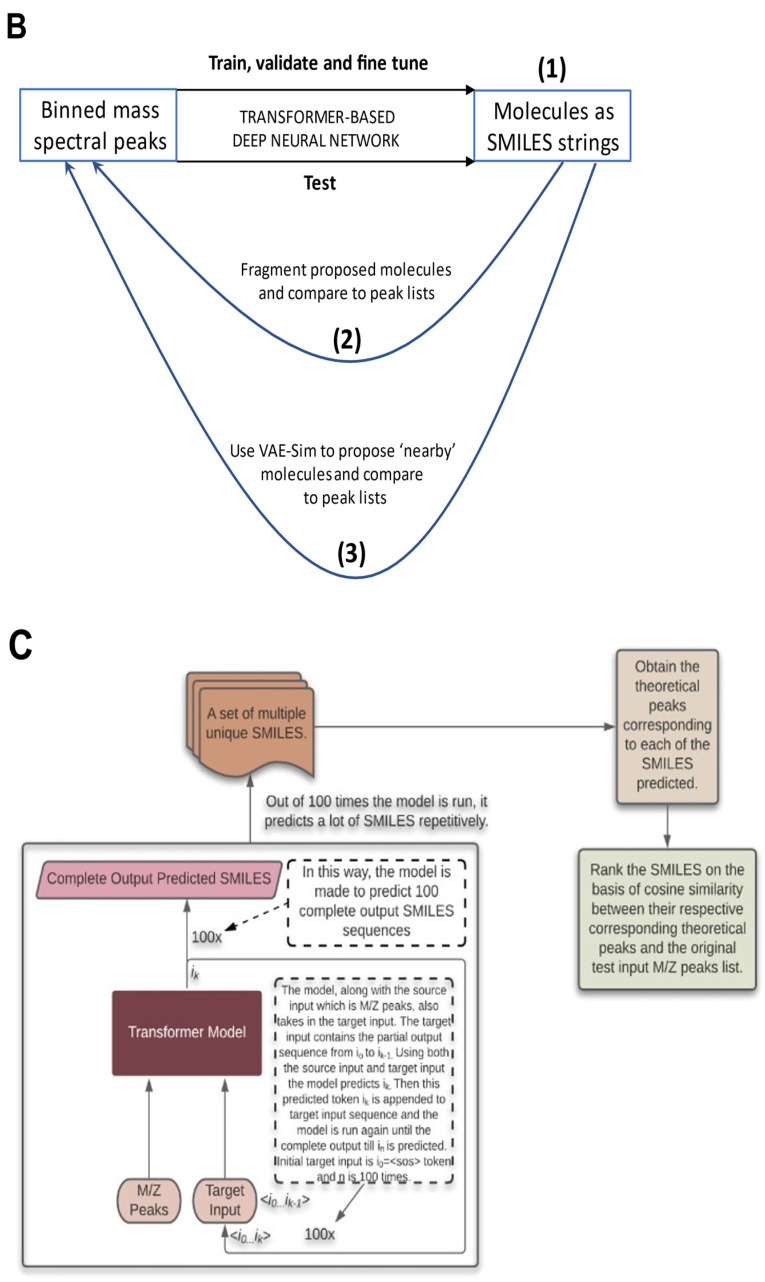
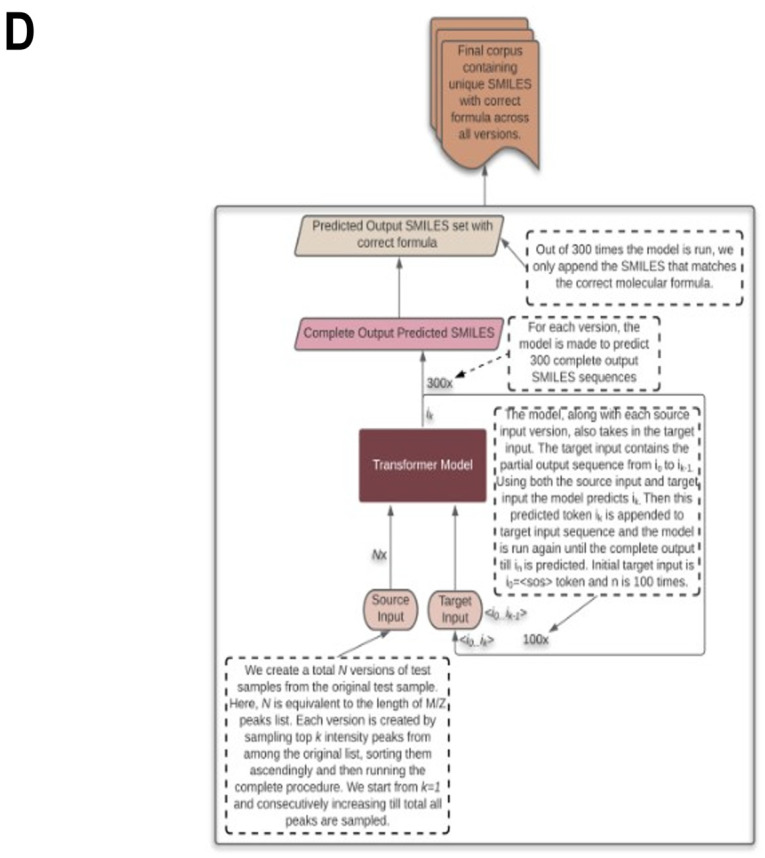
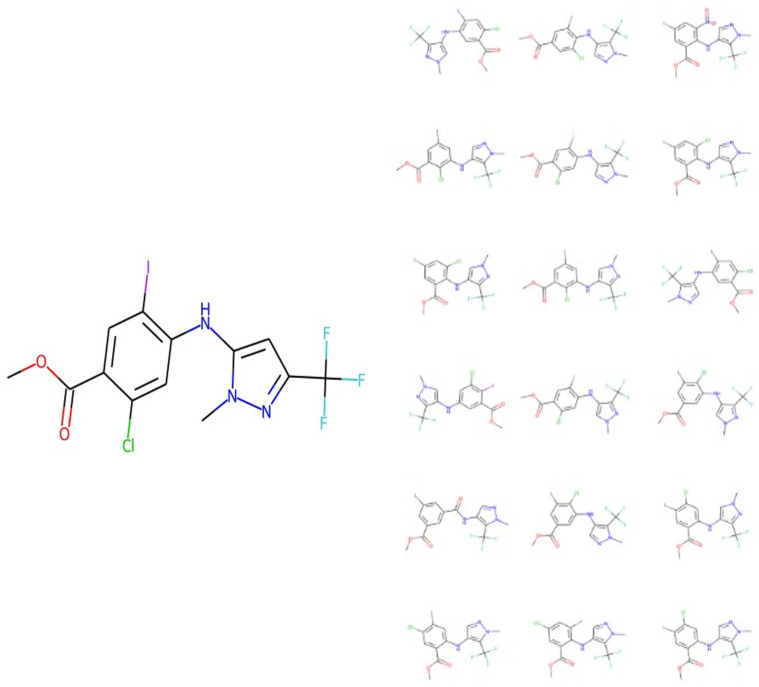

References
-
- Dunn W.B., Broadhurst D., Begley P., Zelena E., Francis-McIntyre S., Anderson N., Brown N., Knowles J., Halsall A., Haselden J.N., et al. The Husermet consortium, Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011;6:1060–1083. doi: 10.1038/nprot.2011.335. - DOI - PubMed
-
- Dunn W.B., Erban A., Weber R.J.M., Creek D.J., Brown M., Breitling R., Hankemeier T., Goodacre R., Neumann S., Kopka J., et al. Mass Appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolites. 2013;9:S44–S66. doi: 10.1007/s11306-012-0434-4. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
