

The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A
- PMID: 34948090
- PMCID: PMC8703989
- DOI: 10.3390/ijms222413294
The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A
Abstract
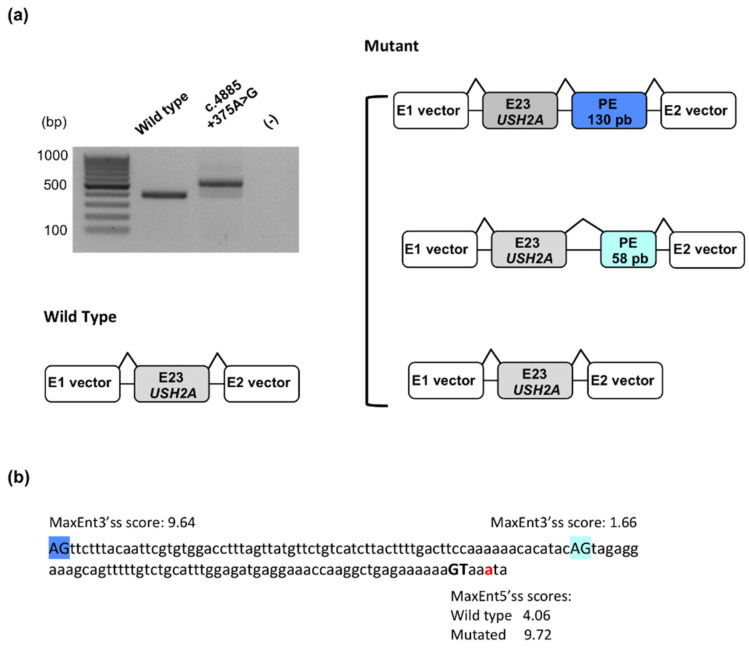
Usher syndrome is an autosomal recessive disorder characterized by congenital hearing loss combined with retinitis pigmentosa, and in some cases, vestibular areflexia. Three clinical subtypes are distinguished, and MYO7A and USH2A represent the two major causal genes involved in Usher type I, the most severe form, and type II, the most frequent form, respectively. Massively parallel sequencing was performed on a cohort of patients in the context of a molecular diagnosis to confirm clinical suspicion of Usher syndrome. We report here 231 pathogenic MYO7A and USH2A genotypes identified in 73 Usher type I and 158 Usher type II patients. Furthermore, we present the ACMG classification of the variants, which comprise all types. Among them, 68 have not been previously reported in the literature, including 12 missense and 16 splice variants. We also report a new deep intronic variant in USH2A. Despite the important number of molecular studies published on these two genes, we show that during the course of routine genetic diagnosis, undescribed variants continue to be identified at a high rate. This is particularly pertinent in the current era, where therapeutic strategies based on DNA or RNA technologies are being developed.
Keywords: ACMG classification; MYO7A; USH2A; Usher syndrome; deep intronic variant; hearing loss; pathogenic genotype; retinitis pigmentosa.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Le Quesne Stabej P., Saihan Z., Rangesh N., Steele-Stallard H.B., Ambrose J., Coffey A., Emmerson J., Haralambous E., Hughes Y., Steel K.P., et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012;49:27–36. doi: 10.1136/jmedgenet-2011-100468. - DOI - PMC - PubMed
-
- Bonnet C., Riahi Z., Chantot-Bastaraud S., Smagghe L., Letexier M., Marcaillou C., Lefèvre G.M., Hardelin J.-P., El-Amraoui A., Singh-Estivalet A., et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016;24:1730–1738. doi: 10.1038/ejhg.2016.99. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
