

Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer's disease-associated genes
- PMID: 34949639
- PMCID: PMC8719885
- DOI: 10.1073/pnas.2112095118
Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer's disease-associated genes
Abstract
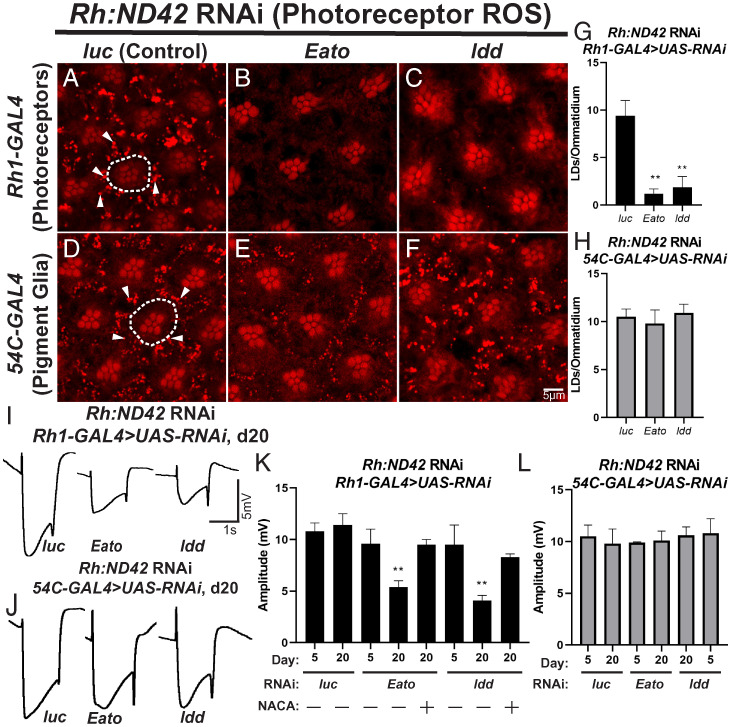
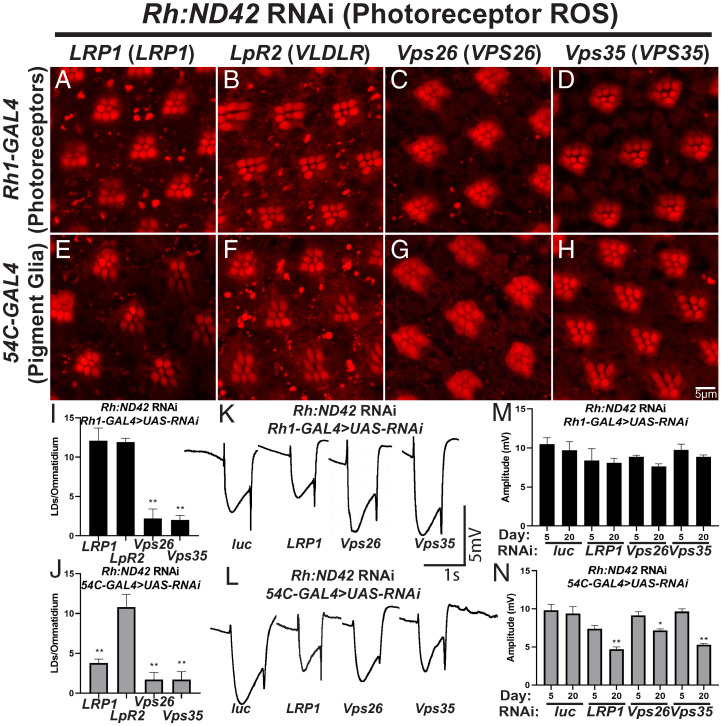
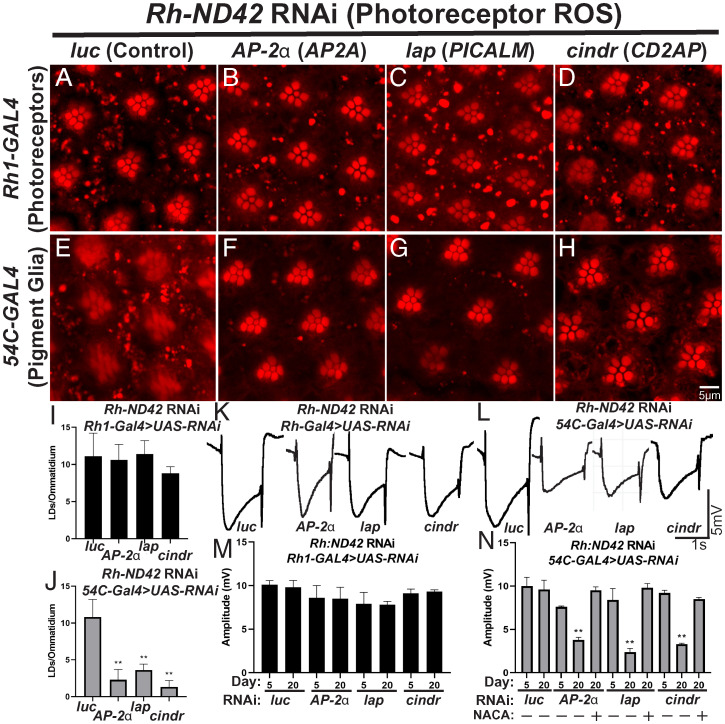
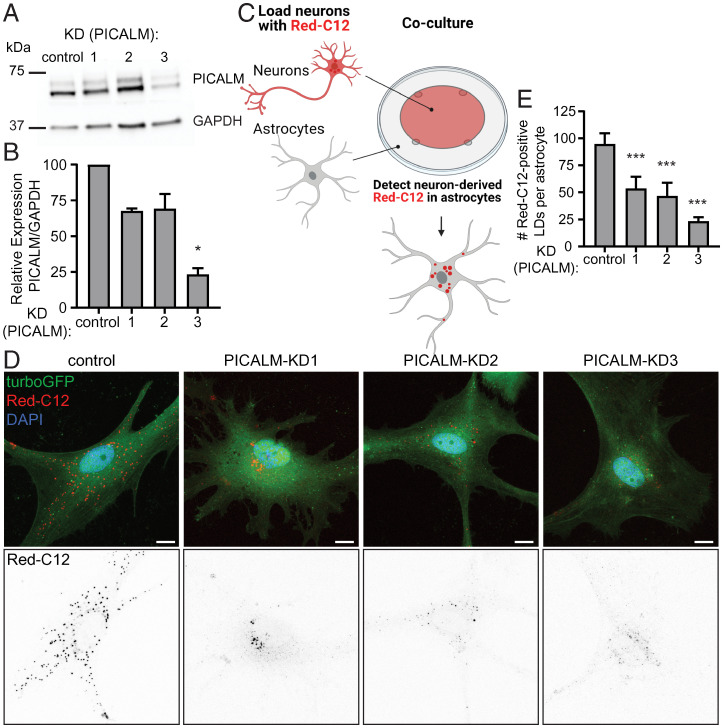
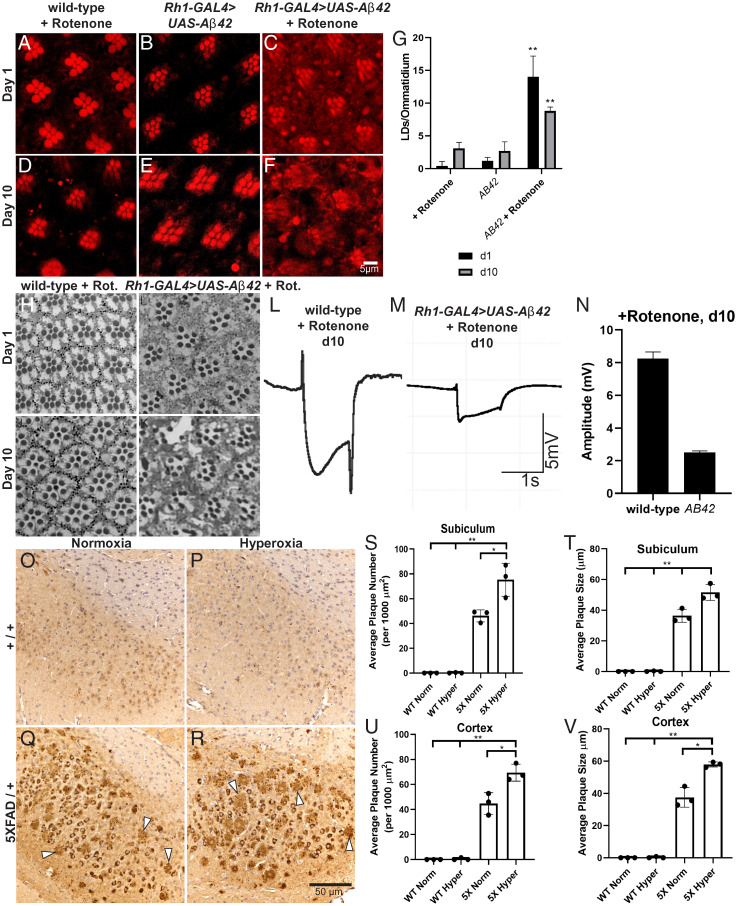
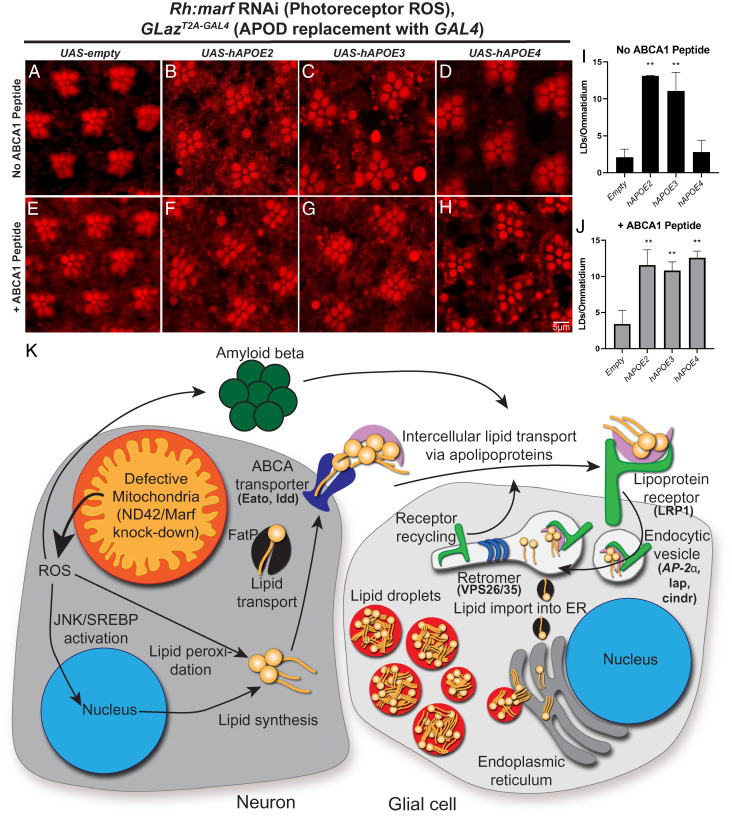
A growing list of Alzheimer's disease (AD) genetic risk factors is being identified, but the contribution of each variant to disease mechanism remains largely unknown. We have previously shown that elevated levels of reactive oxygen species (ROS) induces lipid synthesis in neurons leading to the sequestration of peroxidated lipids in glial lipid droplets (LD), delaying neurotoxicity. This neuron-to-glia lipid transport is APOD/E-dependent. To identify proteins that modulate these neuroprotective effects, we tested the role of AD risk genes in ROS-induced LD formation and demonstrate that several genes impact neuroprotective LD formation, including homologs of human ABCA1, ABCA7, VLDLR, VPS26, VPS35, AP2A, PICALM, and CD2AP Our data also show that ROS enhances Aβ42 phenotypes in flies and mice. Finally, a peptide agonist of ABCA1 restores glial LD formation in a humanized APOE4 fly model, highlighting a potentially therapeutic avenue to prevent ROS-induced neurotoxicity. This study places many AD genetic risk factors in a ROS-induced neuron-to-glia lipid transfer pathway with a critical role in protecting against neurotoxicity.
Keywords: Alzheimer’s disease; Drosophila; GWAS; lipid droplet; peroxidated lipid transfer.
Conflict of interest statement
Competing interest statement: J.O.J. is the President and CEO of Artery Therapeutics, Inc. and part owner of a patent of the peptide used in this work (Patent WO2014144708A1).
Figures
References
-
- Masdeu J. C., Neuroimaging of diseases causing dementia. Neurol. Clin. 38, 65–94 (2020). - PubMed
-
- Bloom G. S., Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508 (2014). - PubMed
-
- De Strooper B., Karran E., The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016). - PubMed
-
- Kunkle B. W., et al. ; Alzheimer Disease Genetics Consortium (ADGC); European Alzheimer’s Disease Initiative (EADI); Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE); Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES), Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
