

Benchmarking atlas-level data integration in single-cell genomics
- PMID: 34949812
- PMCID: PMC8748196
- DOI: 10.1038/s41592-021-01336-8
Benchmarking atlas-level data integration in single-cell genomics
Abstract
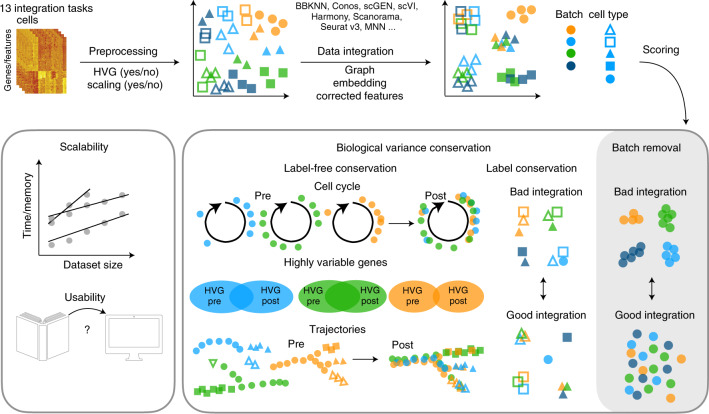
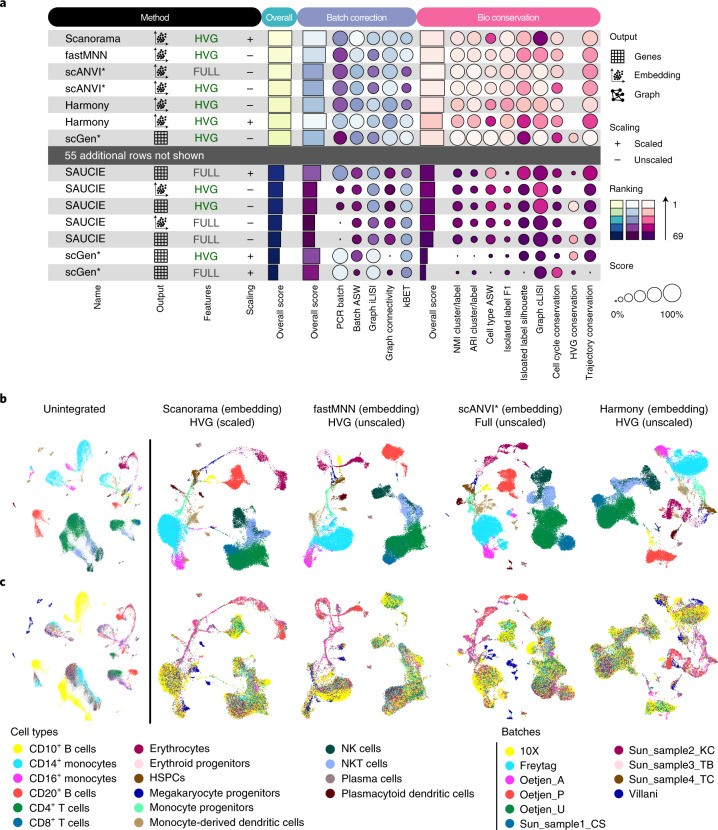
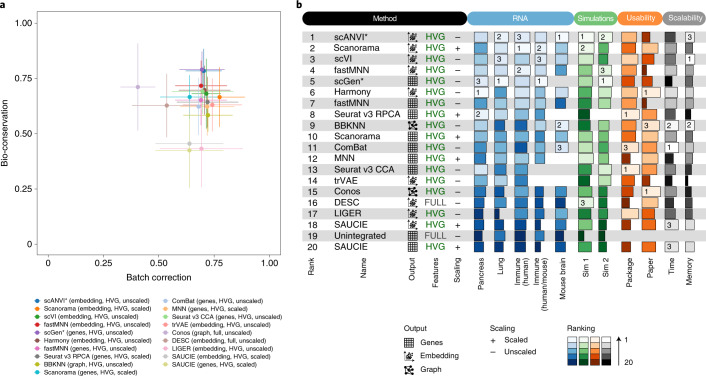
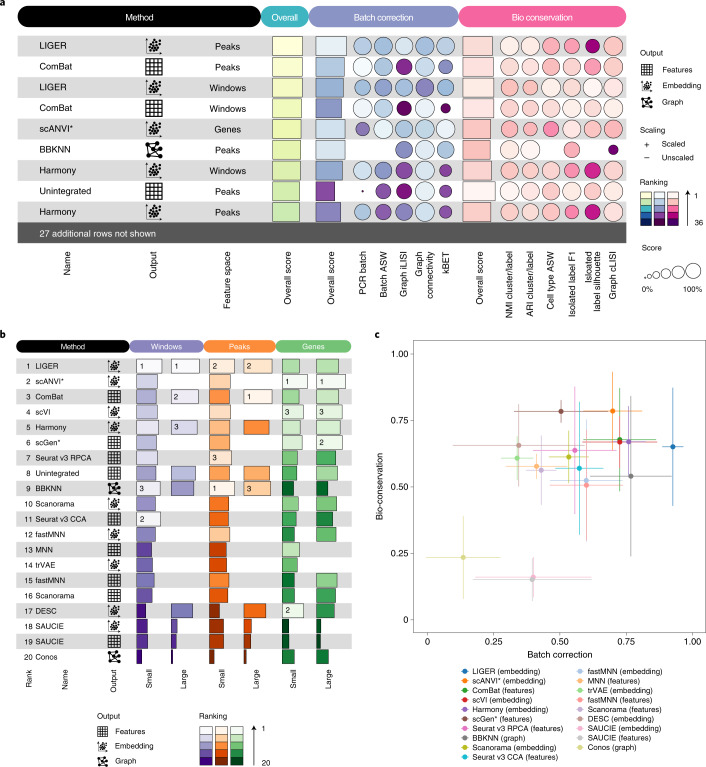
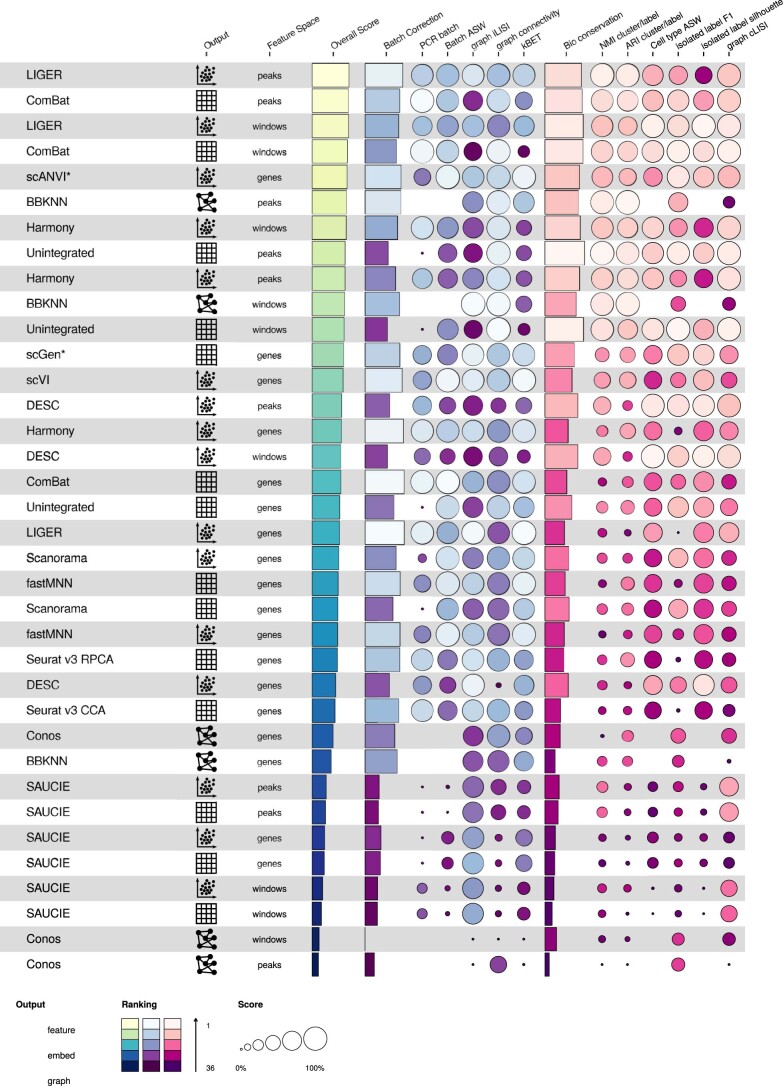
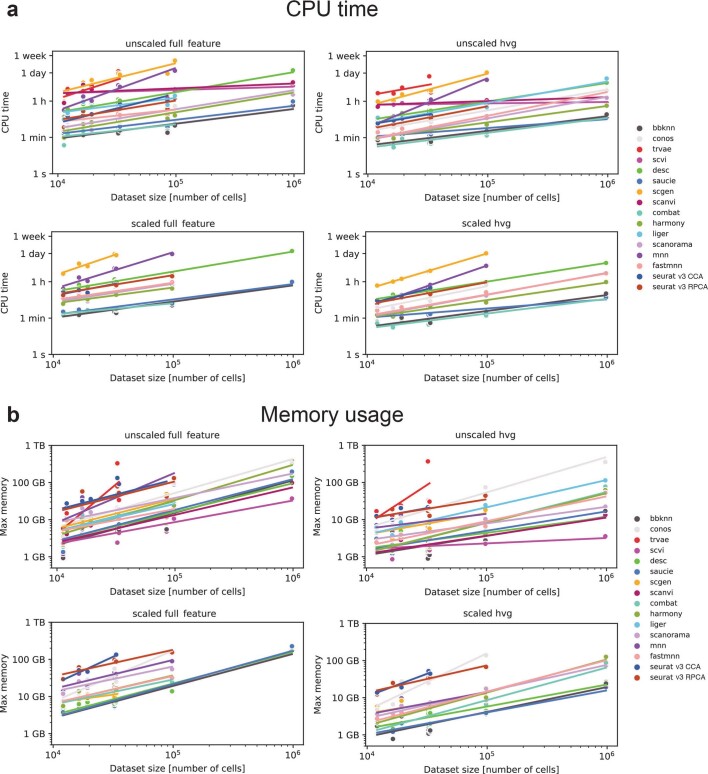
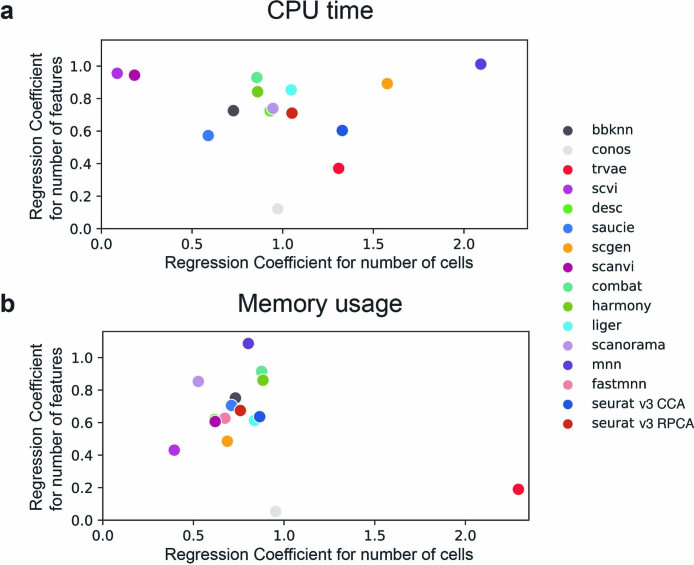
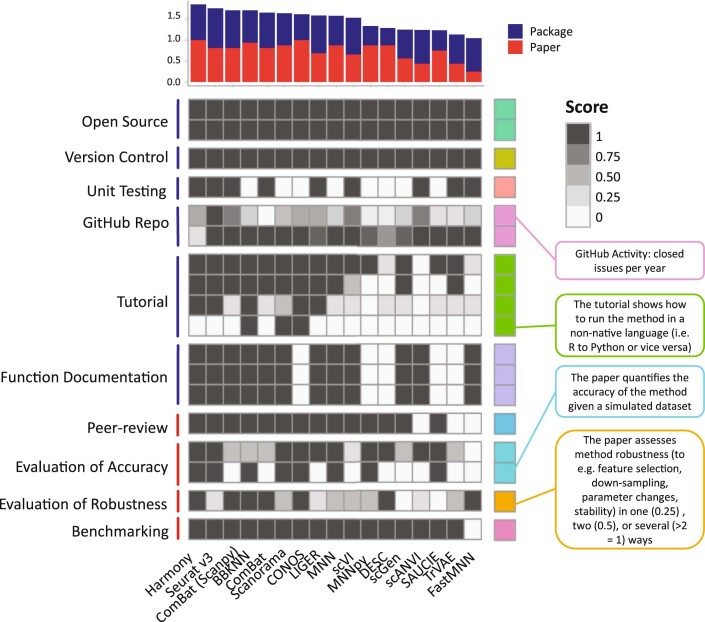
Single-cell atlases often include samples that span locations, laboratories and conditions, leading to complex, nested batch effects in data. Thus, joint analysis of atlas datasets requires reliable data integration. To guide integration method choice, we benchmarked 68 method and preprocessing combinations on 85 batches of gene expression, chromatin accessibility and simulation data from 23 publications, altogether representing >1.2 million cells distributed in 13 atlas-level integration tasks. We evaluated methods according to scalability, usability and their ability to remove batch effects while retaining biological variation using 14 evaluation metrics. We show that highly variable gene selection improves the performance of data integration methods, whereas scaling pushes methods to prioritize batch removal over conservation of biological variation. Overall, scANVI, Scanorama, scVI and scGen perform well, particularly on complex integration tasks, while single-cell ATAC-sequencing integration performance is strongly affected by choice of feature space. Our freely available Python module and benchmarking pipeline can identify optimal data integration methods for new data, benchmark new methods and improve method development.
© 2021. The Author(s).
Conflict of interest statement
F.J.T. reports receiving consulting fees from Immunai and ownership interest in Dermagnostix GmbH and Cellarity. The remaining authors declare no competing interests.
Figures

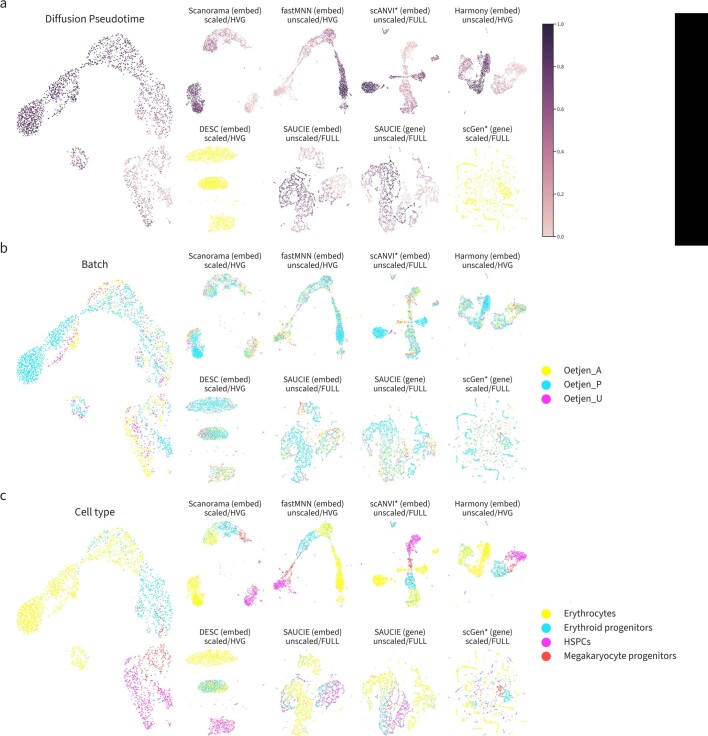
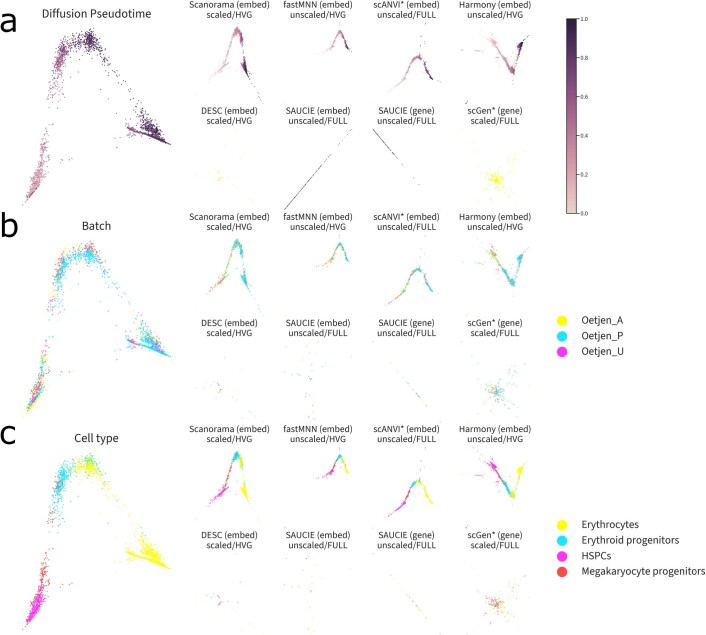
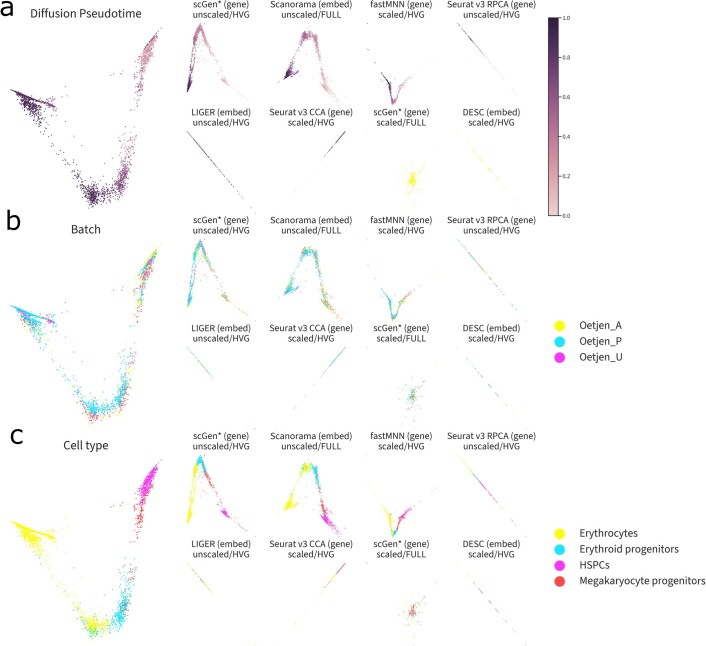
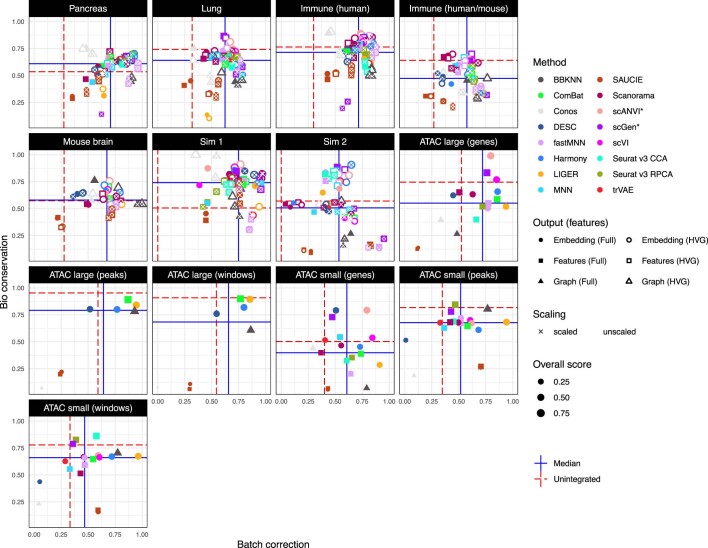

References
-
- Gehring J, Hwee Park J, Chen S, Thomson M, Pachter L. Highly multiplexed single-cell RNA-seq by DNA oligonucleotide tagging of cellular proteins. Nat. Biotechnol. 2020;38:35–38. - PubMed
-
- Mereu E, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020;38:747–755. - PubMed
-
- Regev, A. et al. The Human Cell Atlas white paper. Preprint at 10.7554/eLife.27041 (2018).
-
- Eisenstein M. Single-cell RNA-seq analysis software providers scramble to offer solutions. Nat. Biotechnol. 2020;38:254–257. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
