

Spatial structure governs the mode of tumour evolution
- PMID: 34949822
- PMCID: PMC8825284
- DOI: 10.1038/s41559-021-01615-9
Spatial structure governs the mode of tumour evolution
Abstract
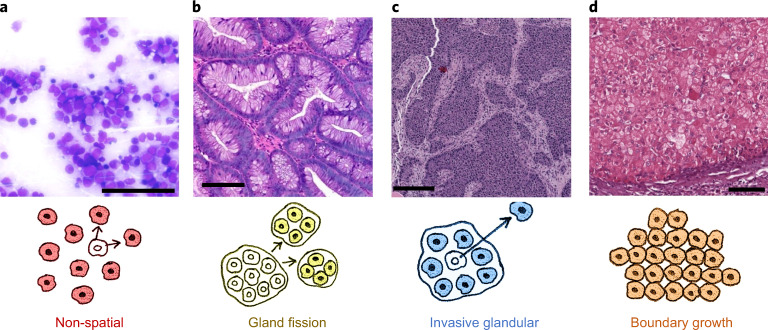
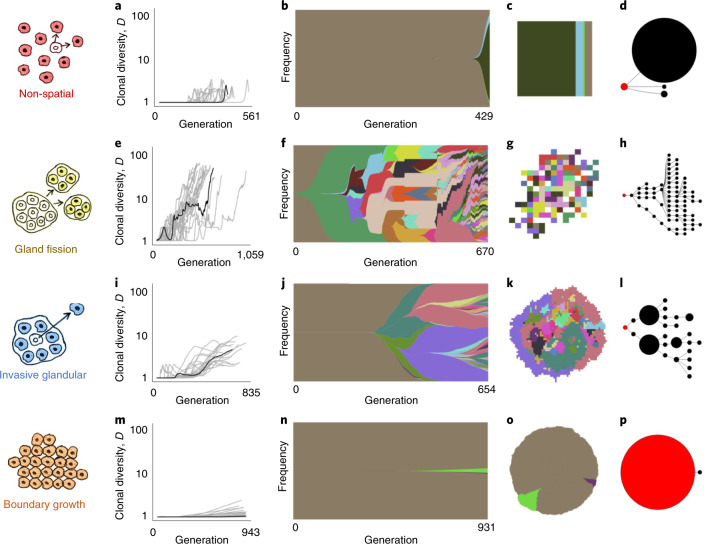
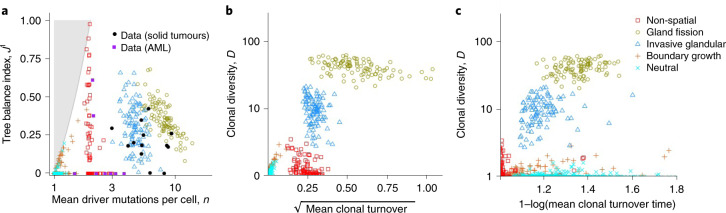
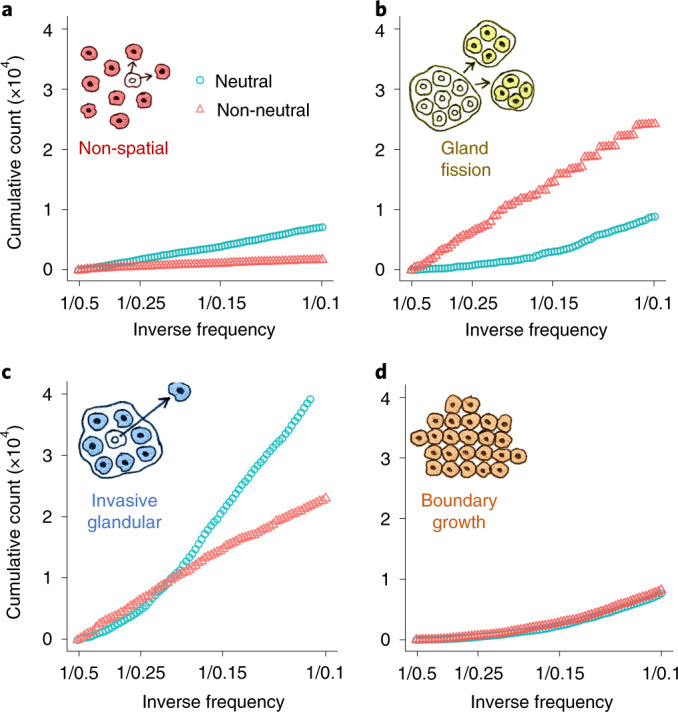
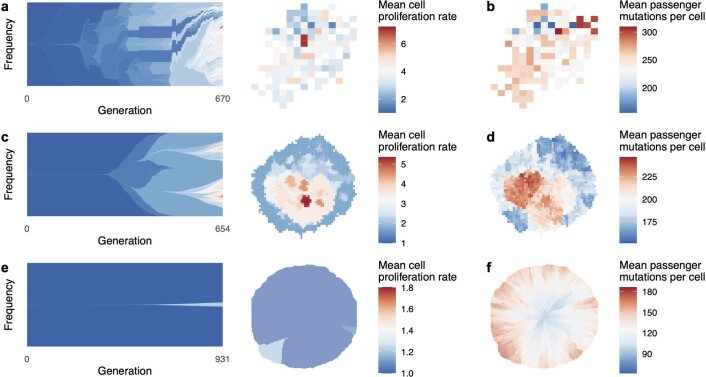
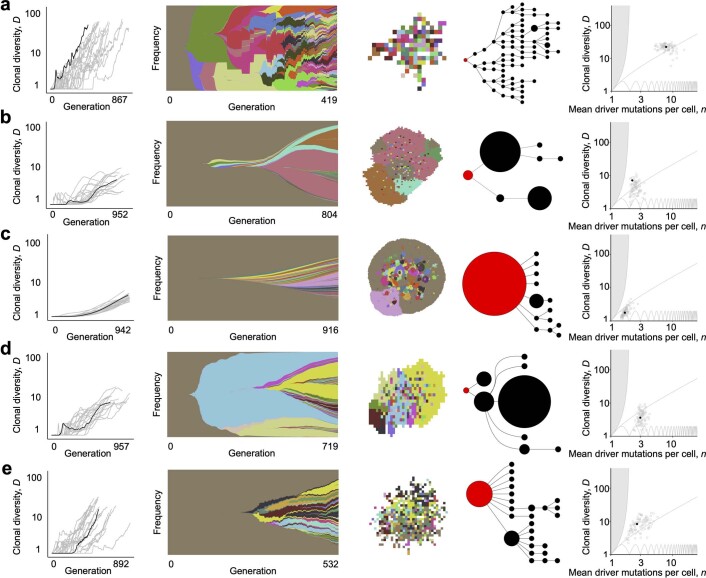
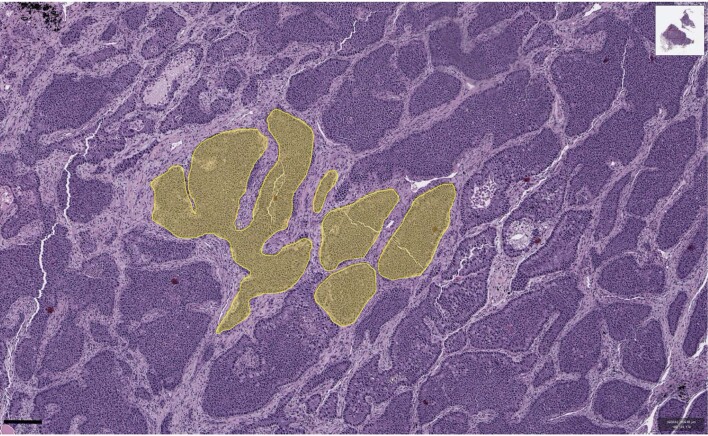
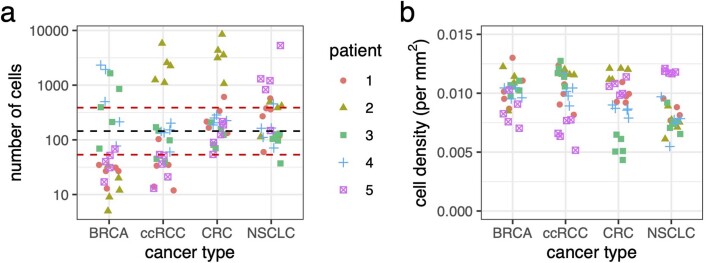
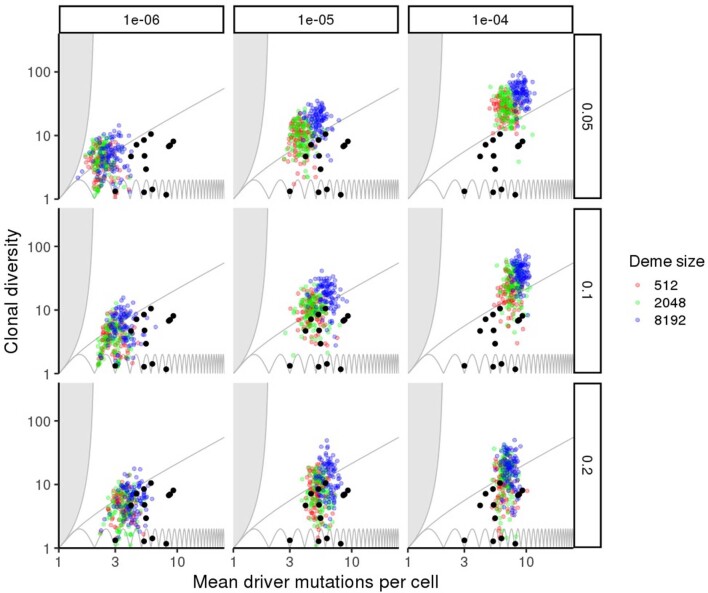
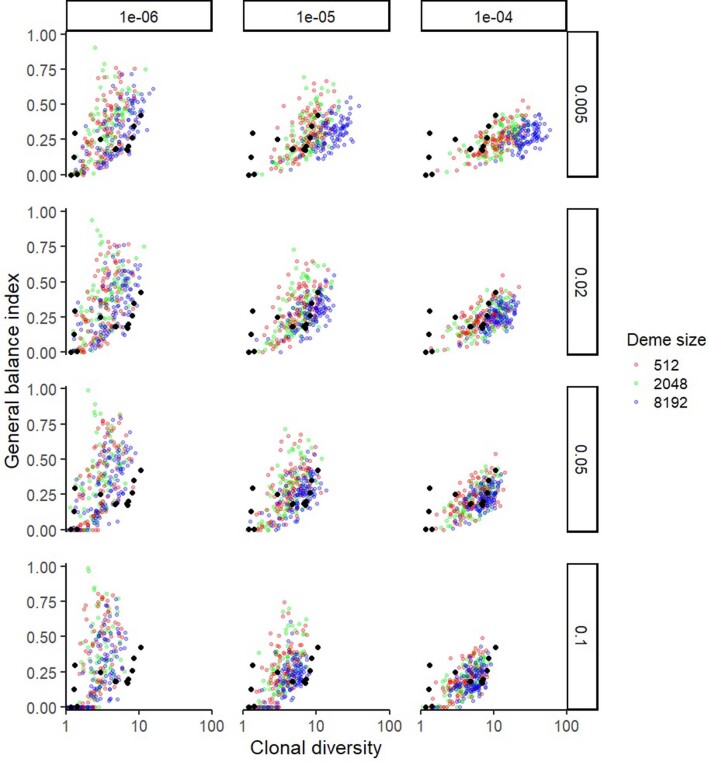
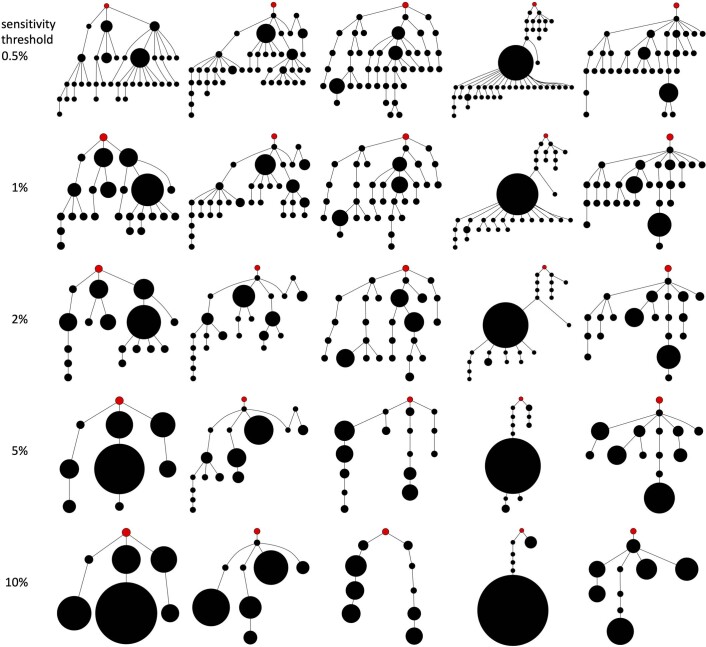
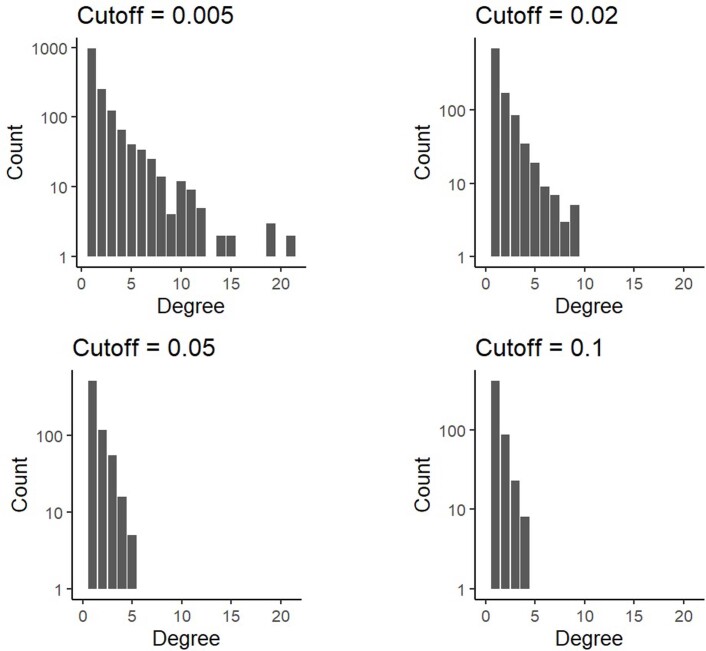
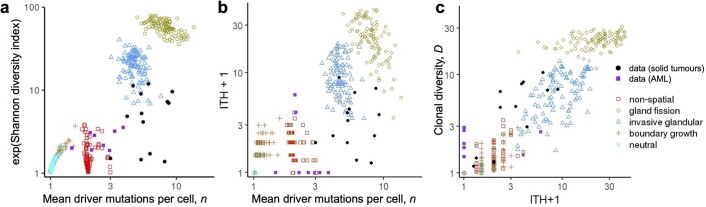
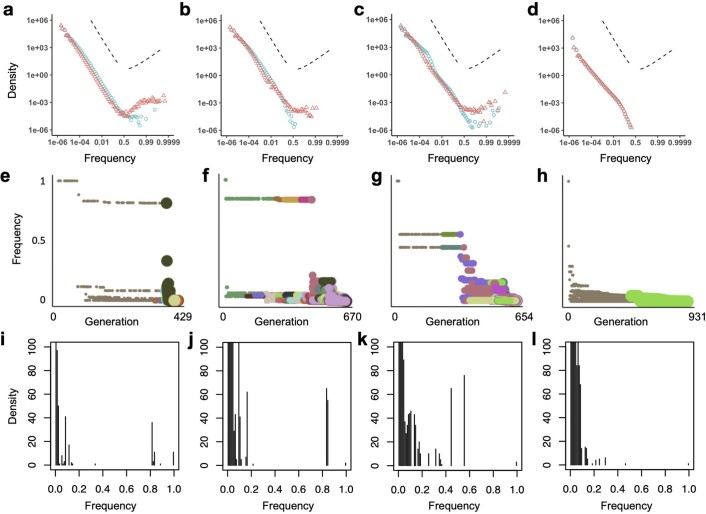
Characterizing the mode-the way, manner or pattern-of evolution in tumours is important for clinical forecasting and optimizing cancer treatment. Sequencing studies have inferred various modes, including branching, punctuated and neutral evolution, but it is unclear why a particular pattern predominates in any given tumour. Here we propose that tumour architecture is key to explaining the variety of observed genetic patterns. We examine this hypothesis using spatially explicit population genetics models and demonstrate that, within biologically relevant parameter ranges, different spatial structures can generate four tumour evolutionary modes: rapid clonal expansion, progressive diversification, branching evolution and effectively almost neutral evolution. Quantitative indices for describing and classifying these evolutionary modes are presented. Using these indices, we show that our model predictions are consistent with empirical observations for cancer types with corresponding spatial structures. The manner of cell dispersal and the range of cell-cell interactions are found to be essential factors in accurately characterizing, forecasting and controlling tumour evolution.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019;20:404–416. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
