

The Identification of a Novel Calcium-Dependent Link Between NAD+ and Glucose Deprivation-Induced Increases in Protein O-GlcNAcylation and ER Stress
- PMID: 34950703
- PMCID: PMC8691773
- DOI: 10.3389/fmolb.2021.780865
The Identification of a Novel Calcium-Dependent Link Between NAD+ and Glucose Deprivation-Induced Increases in Protein O-GlcNAcylation and ER Stress
Abstract
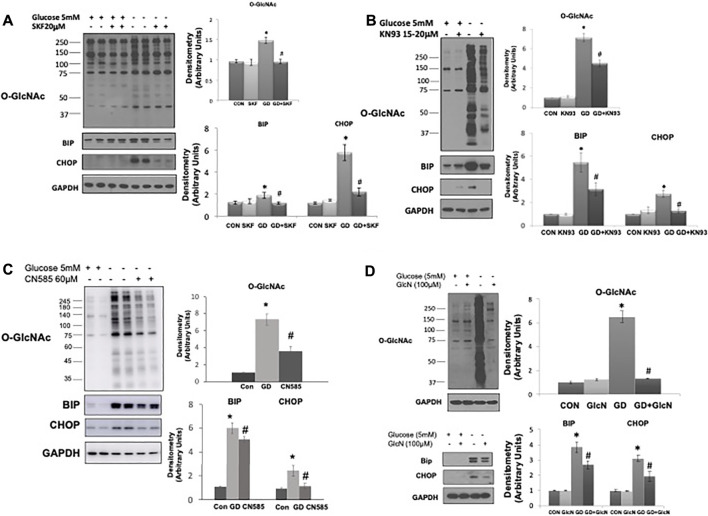
The modification of proteins by O-linked β-N-acetylglucosamine (O-GlcNAc) is associated with the regulation of numerous cellular processes. Despite the importance of O-GlcNAc in mediating cellular function our understanding of the mechanisms that regulate O-GlcNAc levels is limited. One factor known to regulate protein O-GlcNAc levels is nutrient availability; however, the fact that nutrient deficient states such as ischemia increase O-GlcNAc levels suggests that other factors also contribute to regulating O-GlcNAc levels. We have previously reported that in unstressed cardiomyocytes exogenous NAD+ resulted in a time and dose dependent decrease in O-GlcNAc levels. Therefore, we postulated that NAD+ and cellular O-GlcNAc levels may be coordinately regulated. Using glucose deprivation as a model system in an immortalized human ventricular cell line, we examined the influence of extracellular NAD+ on cellular O-GlcNAc levels and ER stress in the presence and absence of glucose. We found that NAD+ completely blocked the increase in O-GlcNAc induced by glucose deprivation and suppressed the activation of ER stress. The NAD+ metabolite cyclic ADP-ribose (cADPR) had similar effects on O-GlcNAc and ER stress suggesting a common underlying mechanism. cADPR is a ryanodine receptor (RyR) agonist and like caffeine, which also activates the RyR, both mimicked the effects of NAD+. SERCA inhibition, which also reduces ER/SR Ca2+ levels had similar effects to both NAD+ and cADPR on O-GlcNAc and ER stress responses to glucose deprivation. The observation that NAD+, cADPR, and caffeine all attenuated the increase in O-GlcNAc and ER stress in response to glucose deprivation, suggests a potential common mechanism, linked to ER/SR Ca2+ levels, underlying their activation. Moreover, we showed that TRPM2, a plasma membrane cation channel was necessary for the cellular responses to glucose deprivation. Collectively, these findings support a novel Ca2+-dependent mechanism underlying glucose deprivation induced increase in O-GlcNAc and ER stress.
Keywords: ER stress; NAD+; O-GlcNAc; TRPM2 cation channel; calcium; glucose deprivation.
Copyright © 2021 Zou, Collins, Young, Zhang, Wende, Darley-Usmar and Chatham.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous