

A dual endosymbiosis supports nutritional adaptation to hematophagy in the invasive tick Hyalomma marginatum
- PMID: 34951405
- PMCID: PMC8709577
- DOI: 10.7554/eLife.72747
A dual endosymbiosis supports nutritional adaptation to hematophagy in the invasive tick Hyalomma marginatum
Abstract
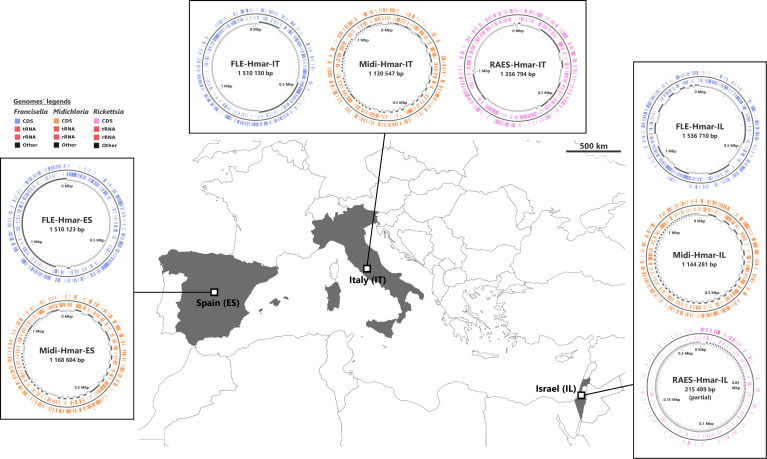
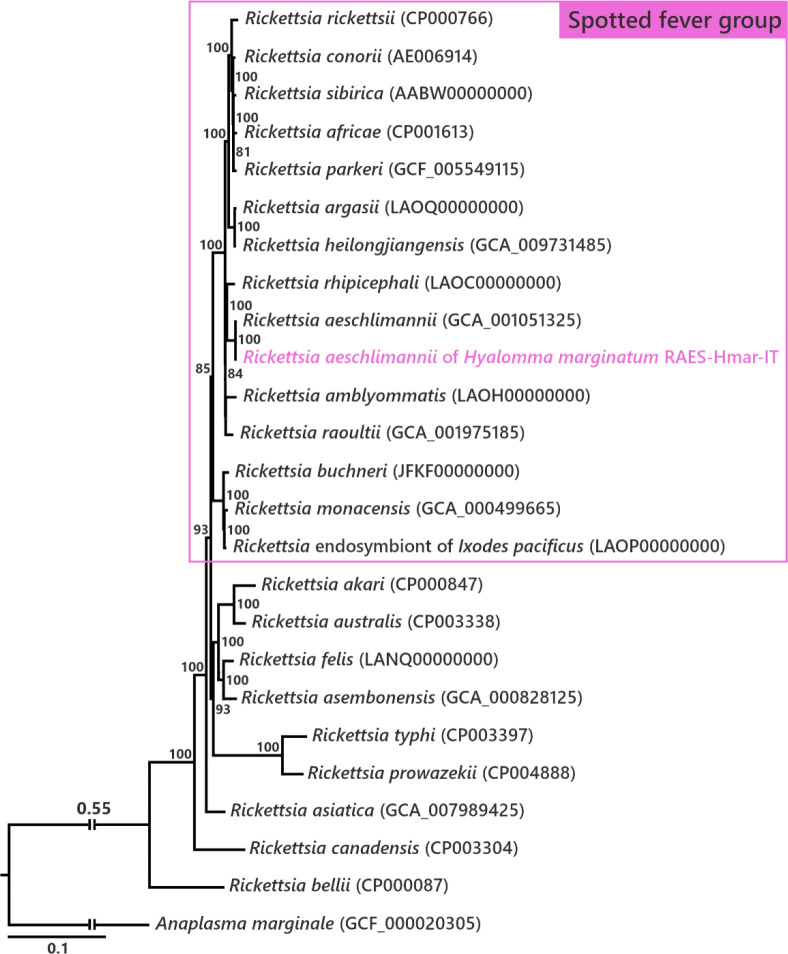
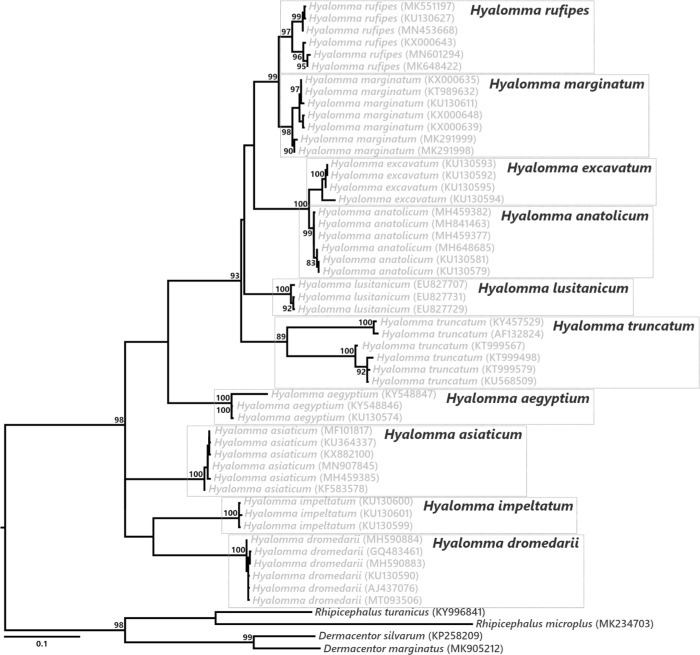
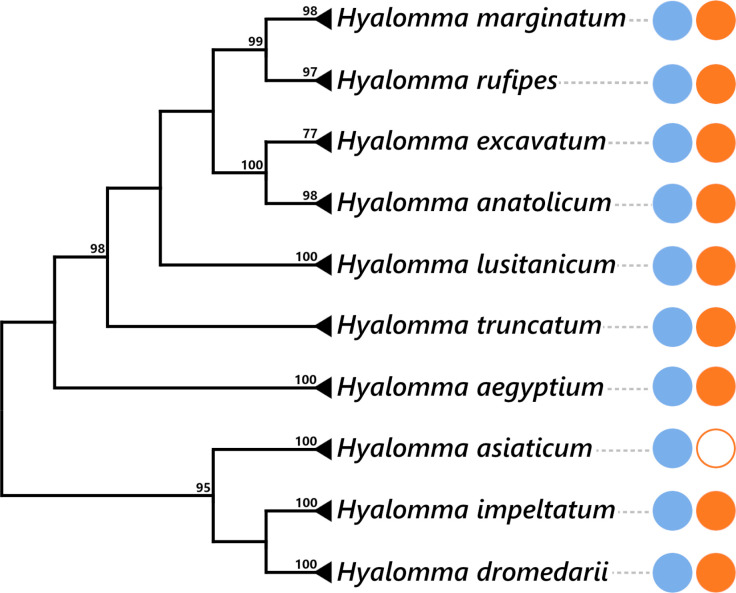
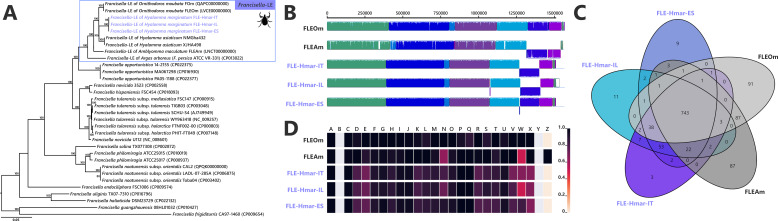
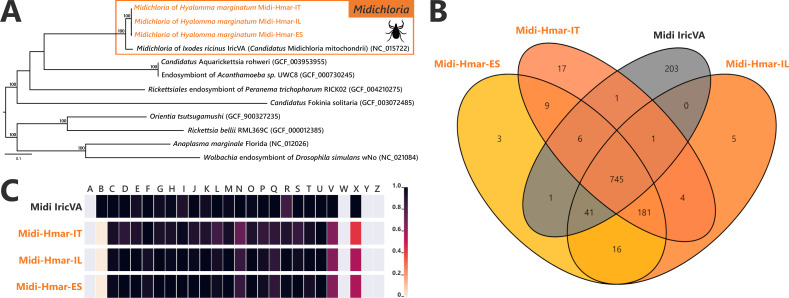
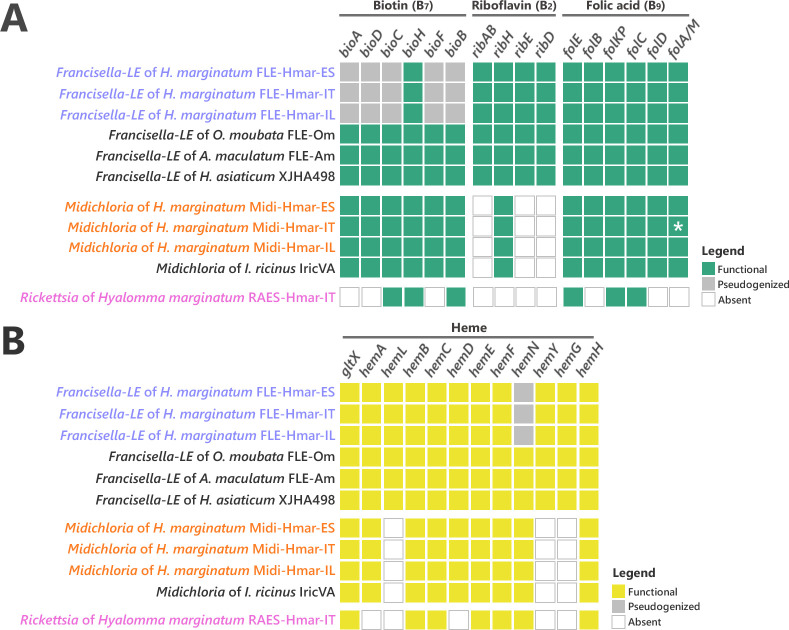
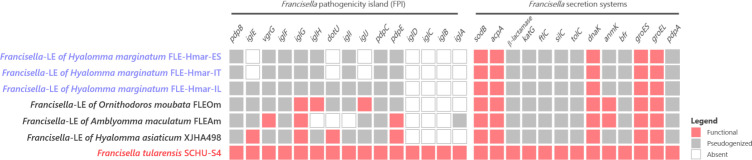
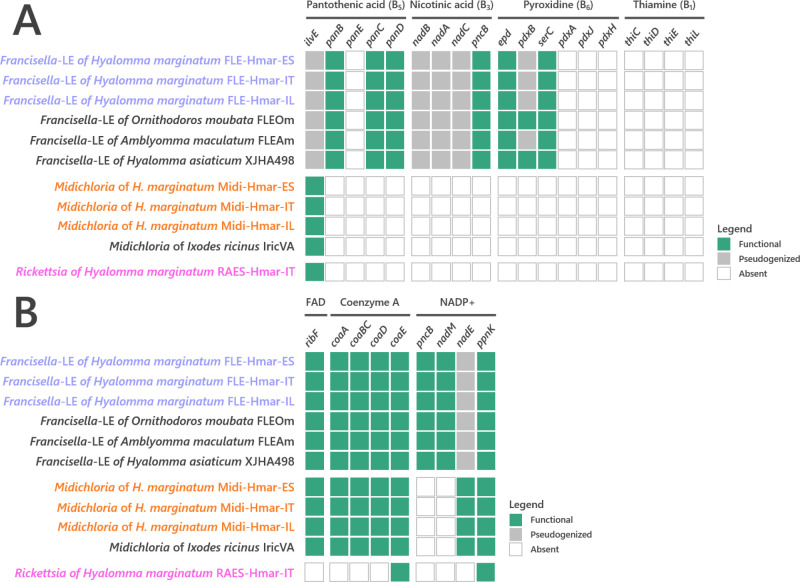
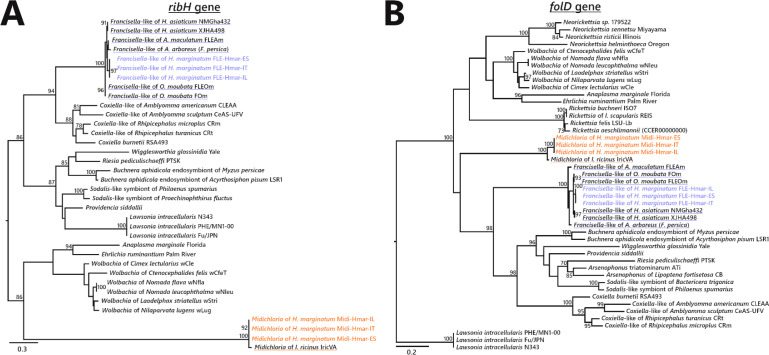
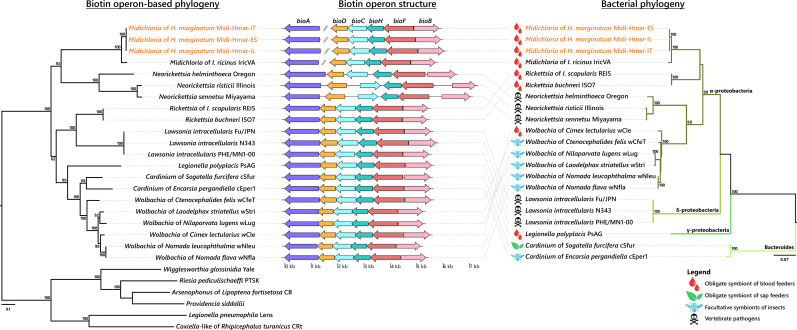
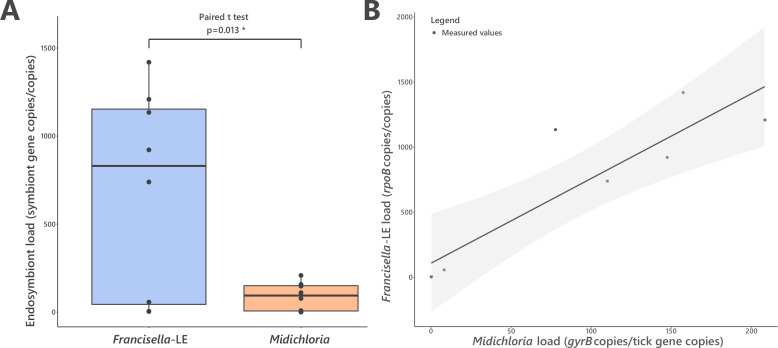
Many animals are dependent on microbial partners that provide essential nutrients lacking from their diet. Ticks, whose diet consists exclusively on vertebrate blood, rely on maternally inherited bacterial symbionts to supply B vitamins. While previously studied tick species consistently harbor a single lineage of those nutritional symbionts, we evidence here that the invasive tick Hyalomma marginatum harbors a unique dual-partner nutritional system between an ancestral symbiont, Francisella, and a more recently acquired symbiont, Midichloria. Using metagenomics, we show that Francisella exhibits extensive genome erosion that endangers the nutritional symbiotic interactions. Its genome includes folate and riboflavin biosynthesis pathways but deprived functional biotin biosynthesis on account of massive pseudogenization. Co-symbiosis compensates this deficiency since the Midichloria genome encompasses an intact biotin operon, which was primarily acquired via lateral gene transfer from unrelated intracellular bacteria commonly infecting arthropods. Thus, in H. marginatum, a mosaic of co-evolved symbionts incorporating gene combinations of distant phylogenetic origins emerged to prevent the collapse of an ancestral nutritional symbiosis. Such dual endosymbiosis was never reported in other blood feeders but was recently documented in agricultural pests feeding on plant sap, suggesting that it may be a key mechanism for advanced adaptation of arthropods to specialized diets.
Keywords: Francisella; Hyalomma; Midichloria; endosymbiosis; evolutionary biology; hematophagy.
© 2021, Buysse et al.
Conflict of interest statement
MB, AF, YG, TN, FC, EO, AG, AP, BM, CB, AC, DS, OD No competing interests declared
Figures


References
-
- Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. ScienceOpen, Inc; 2010.
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
