

Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions
- PMID: 34952941
- PMCID: PMC8732310
- DOI: 10.1038/s41564-021-01011-w
Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions
Abstract
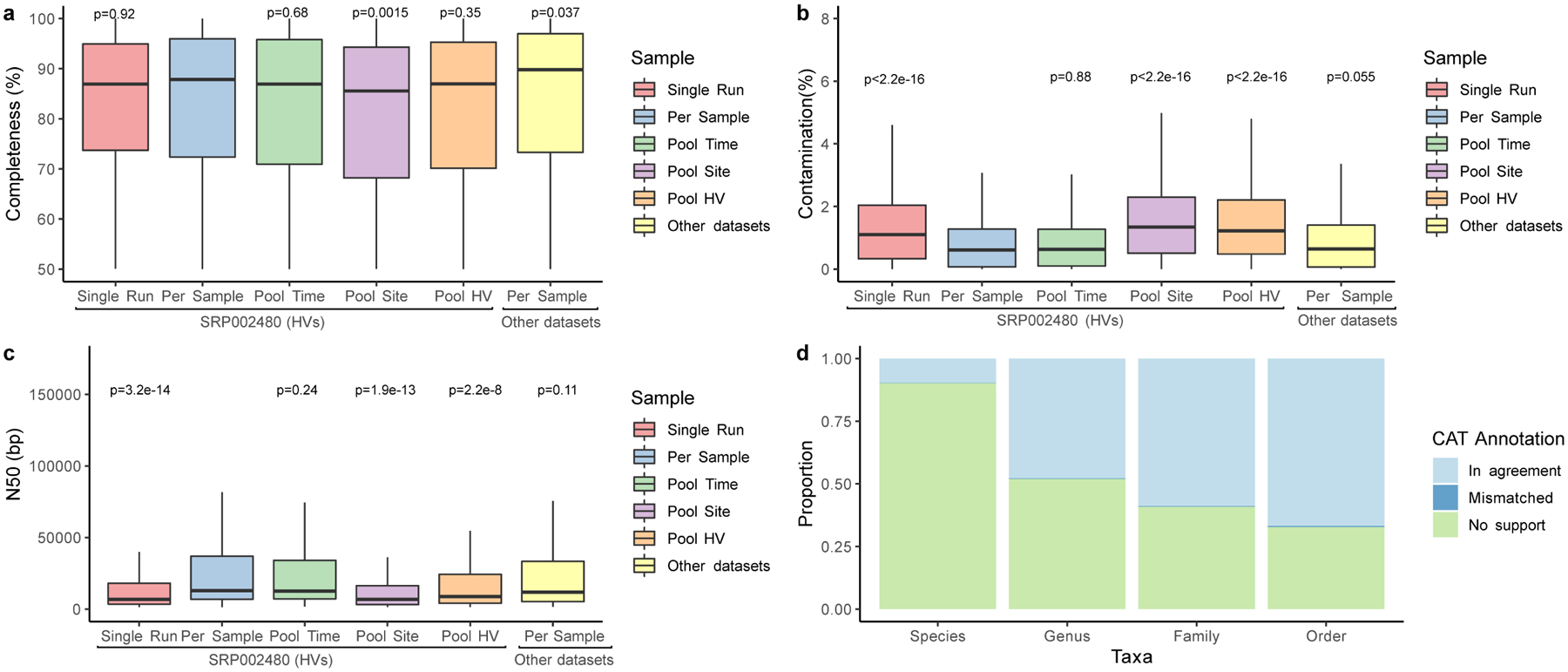
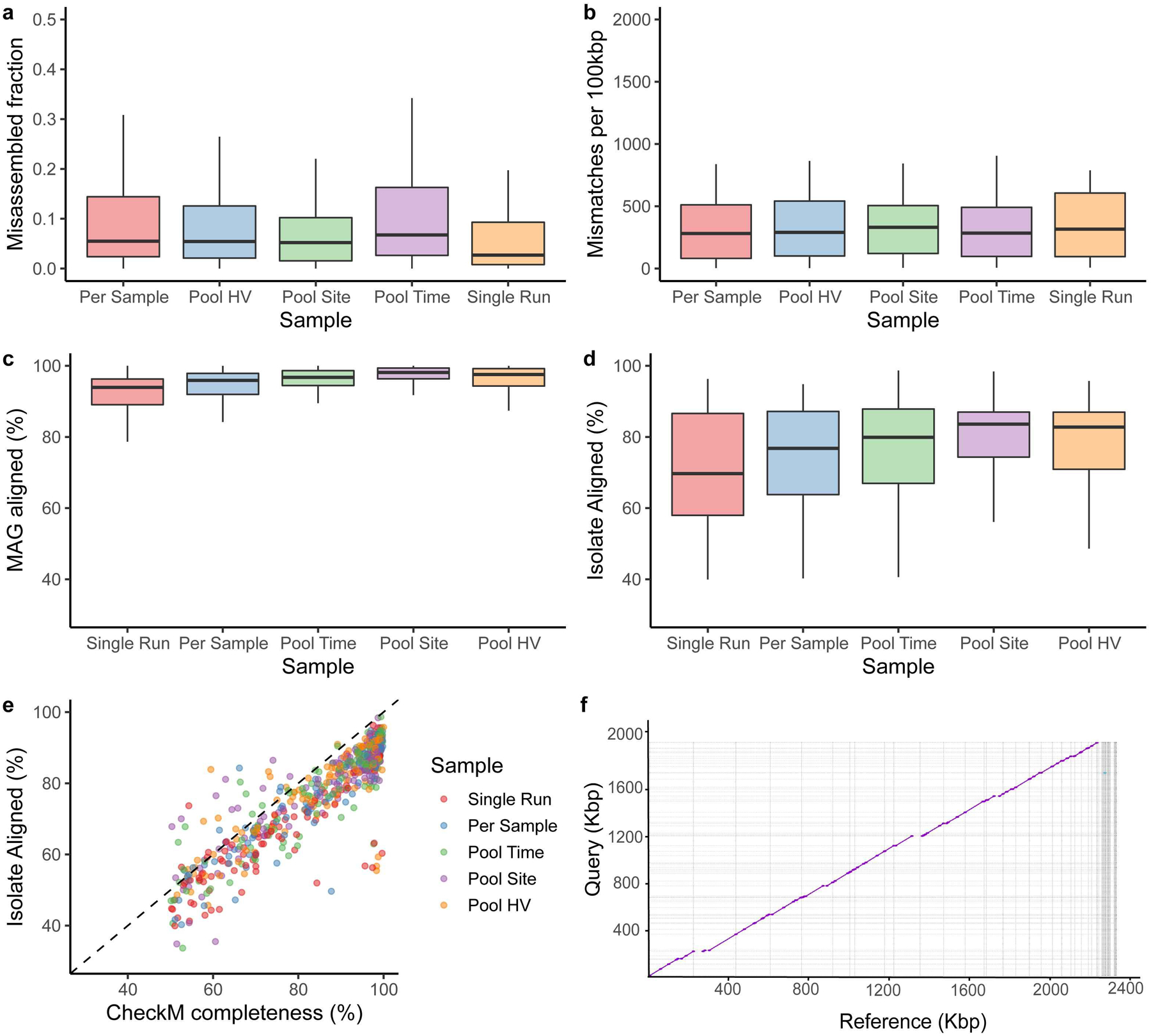
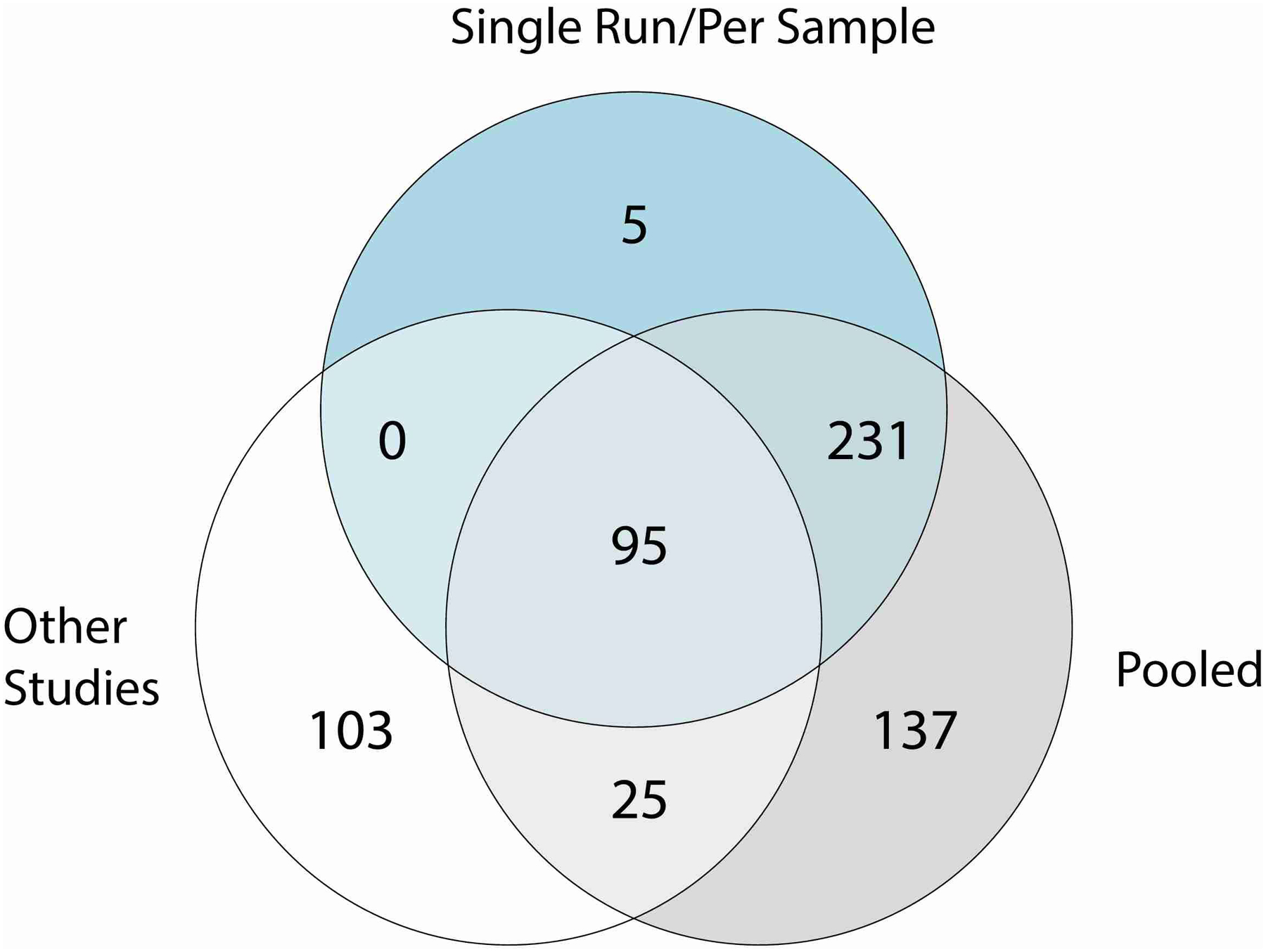
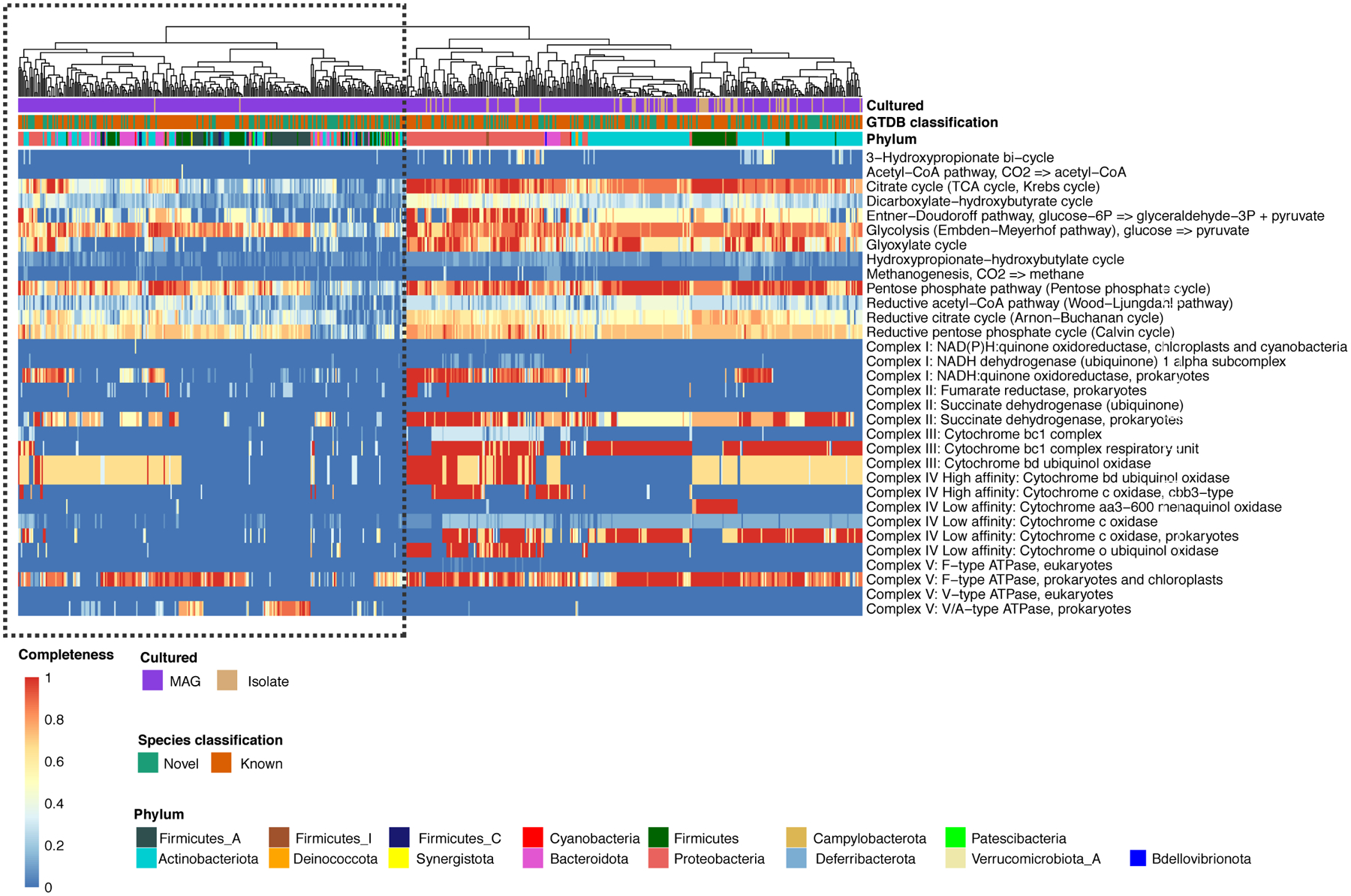
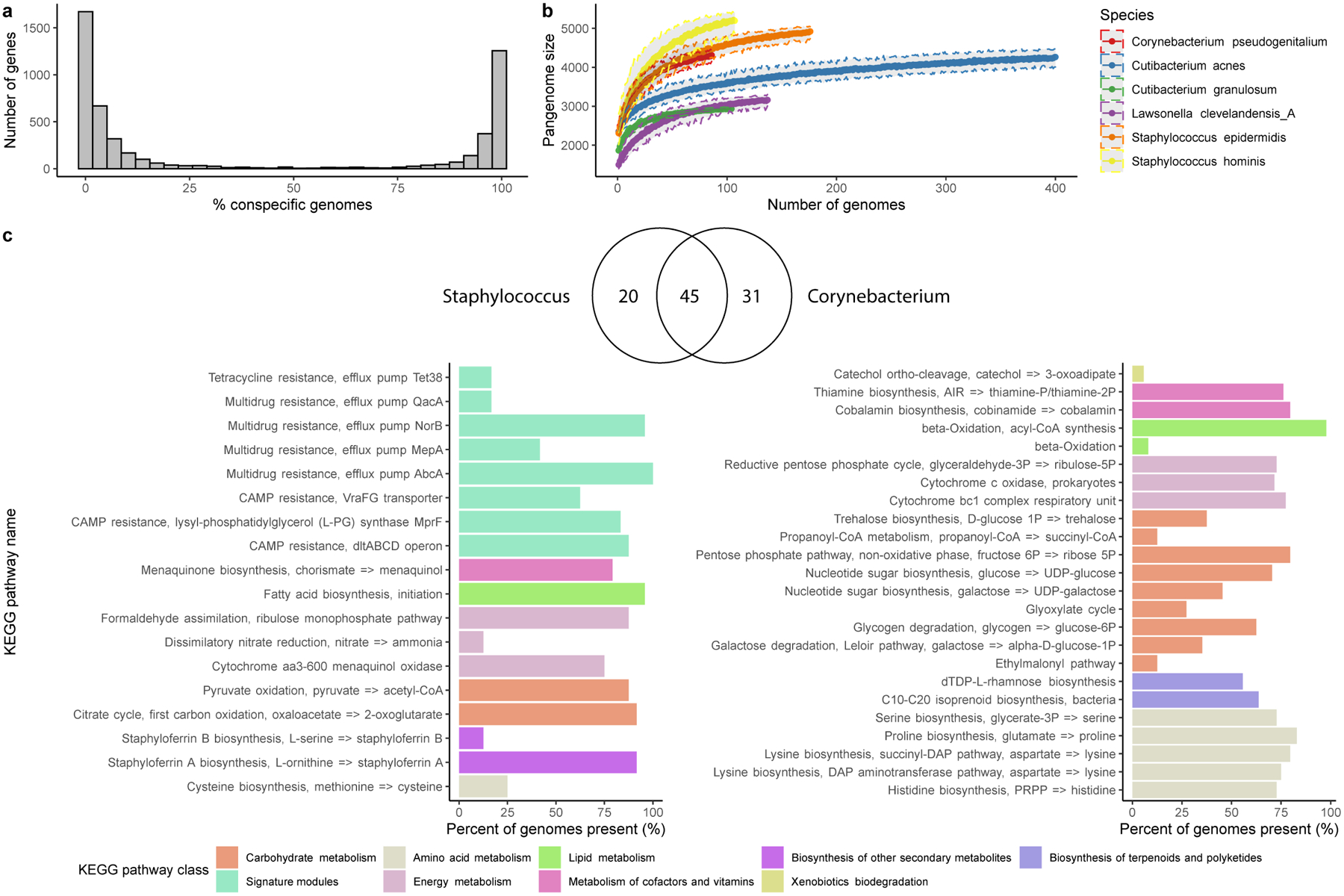
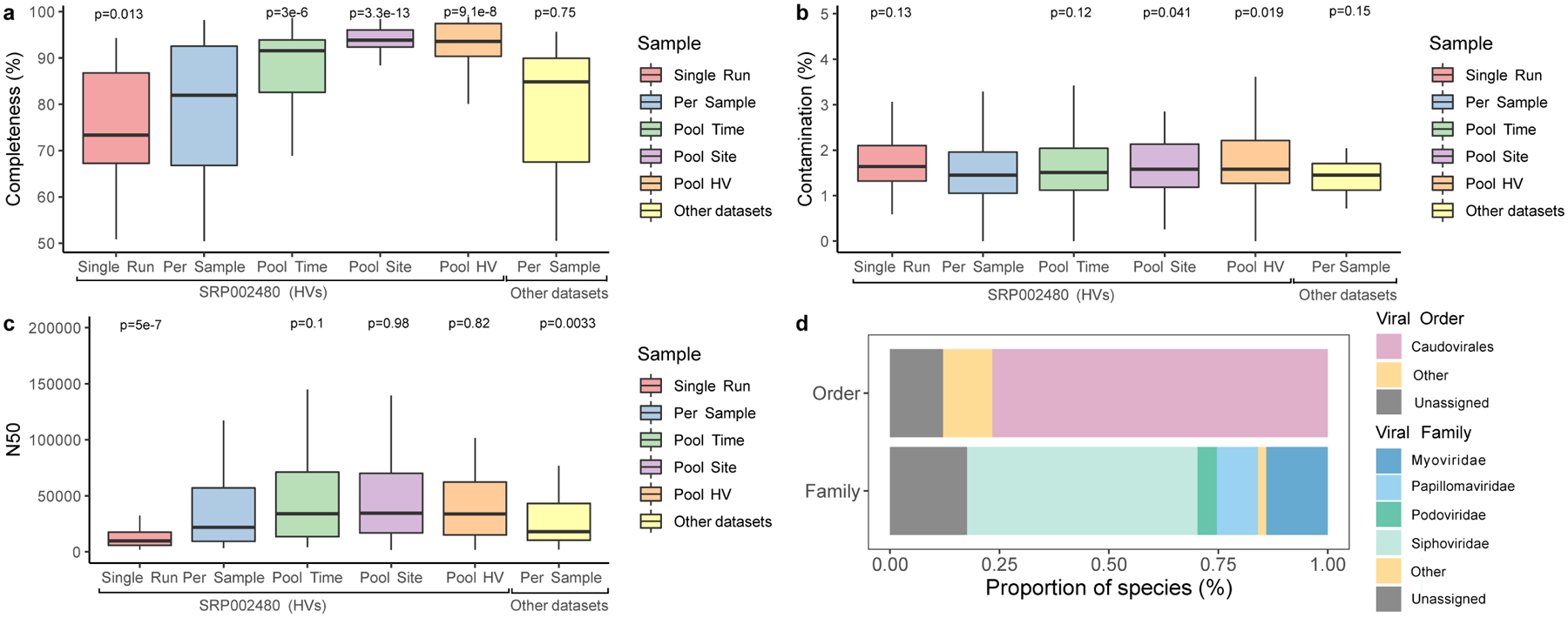
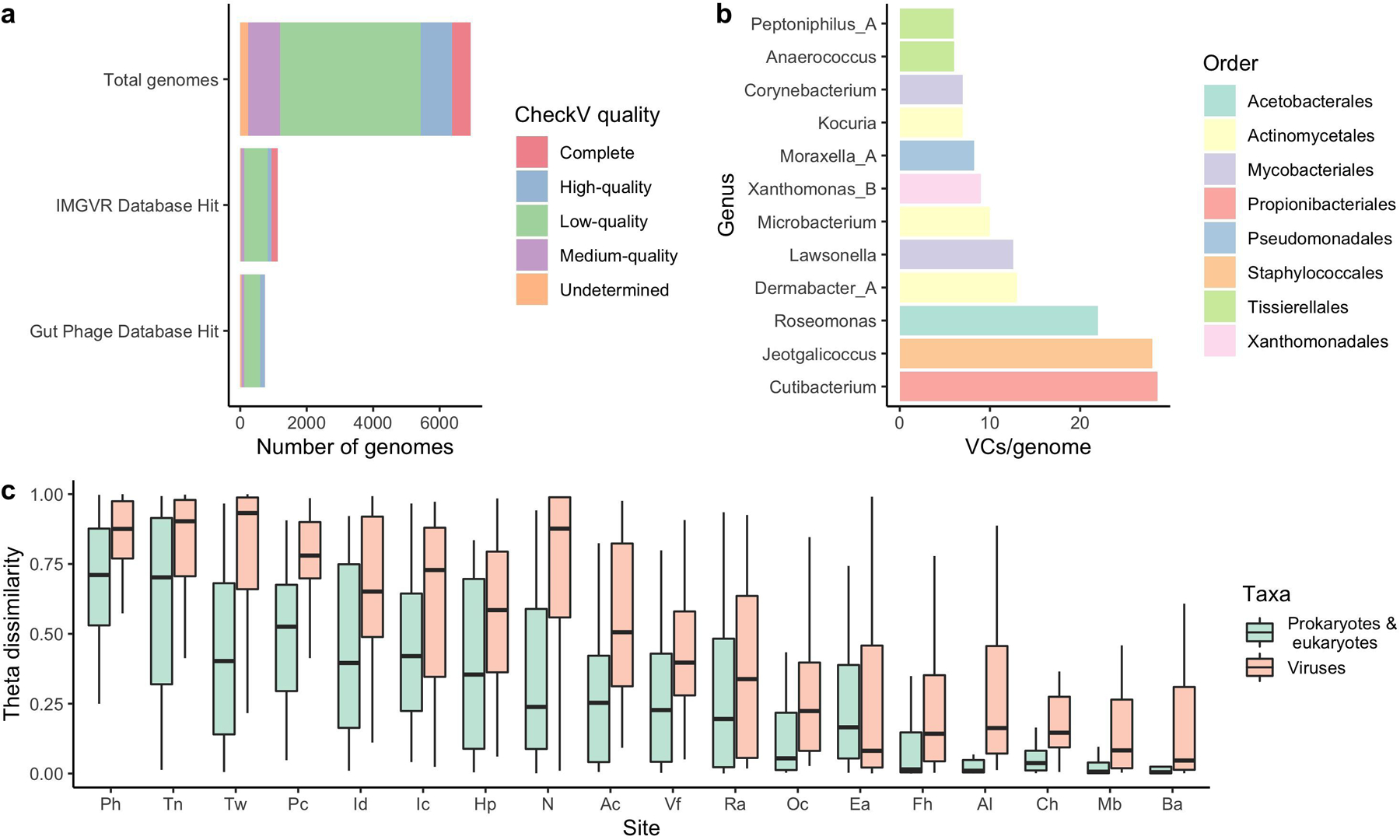
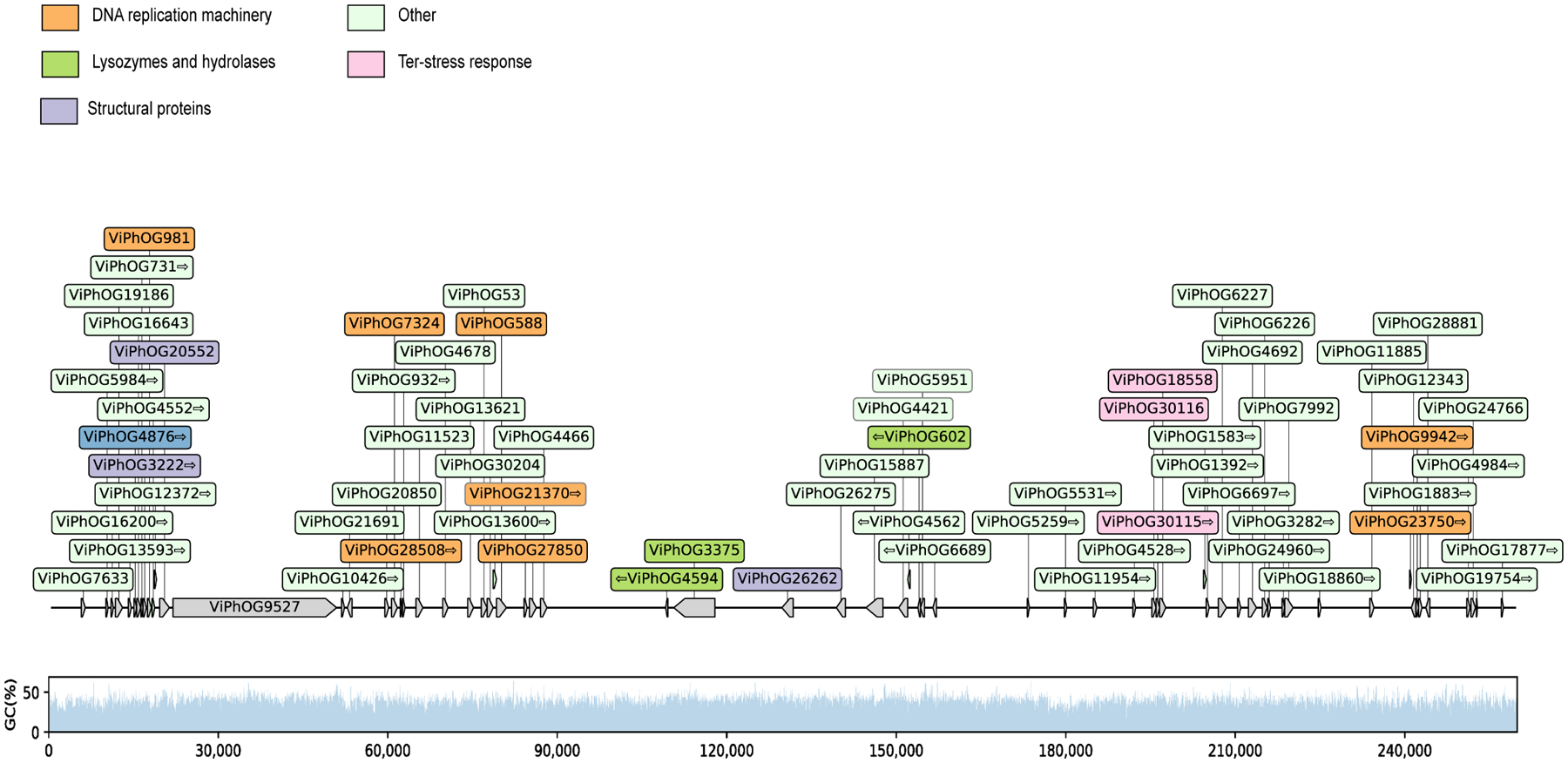
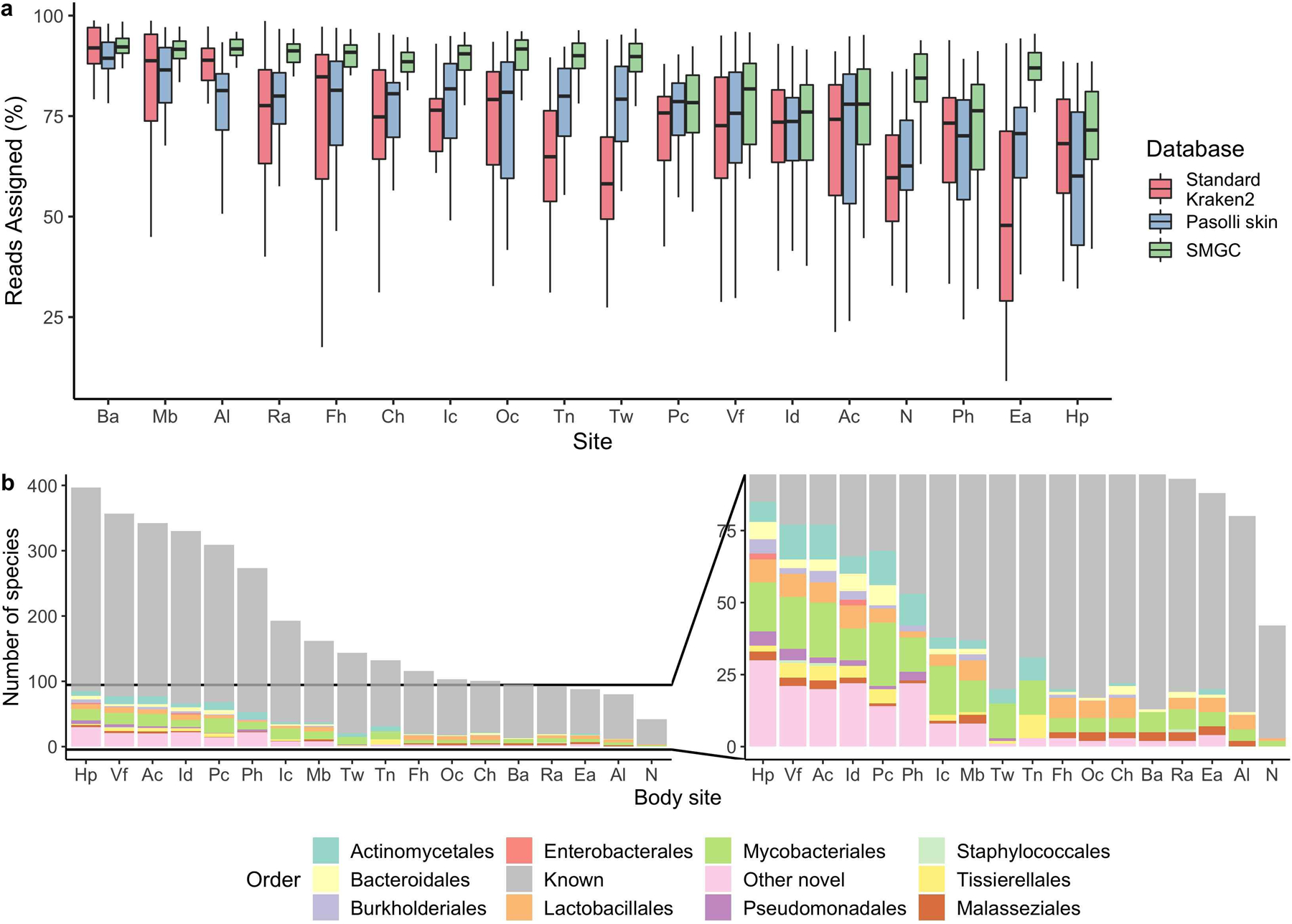
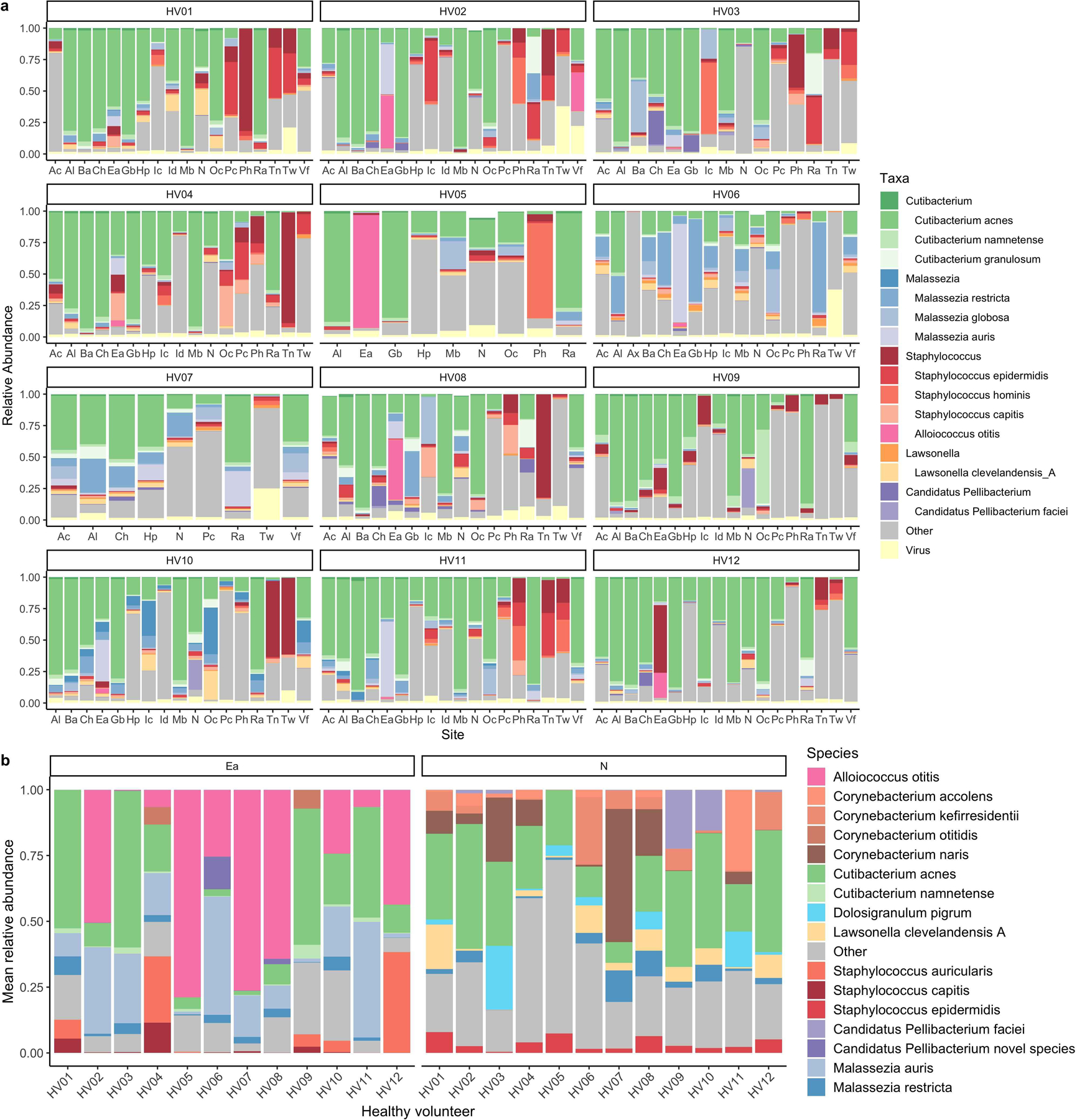
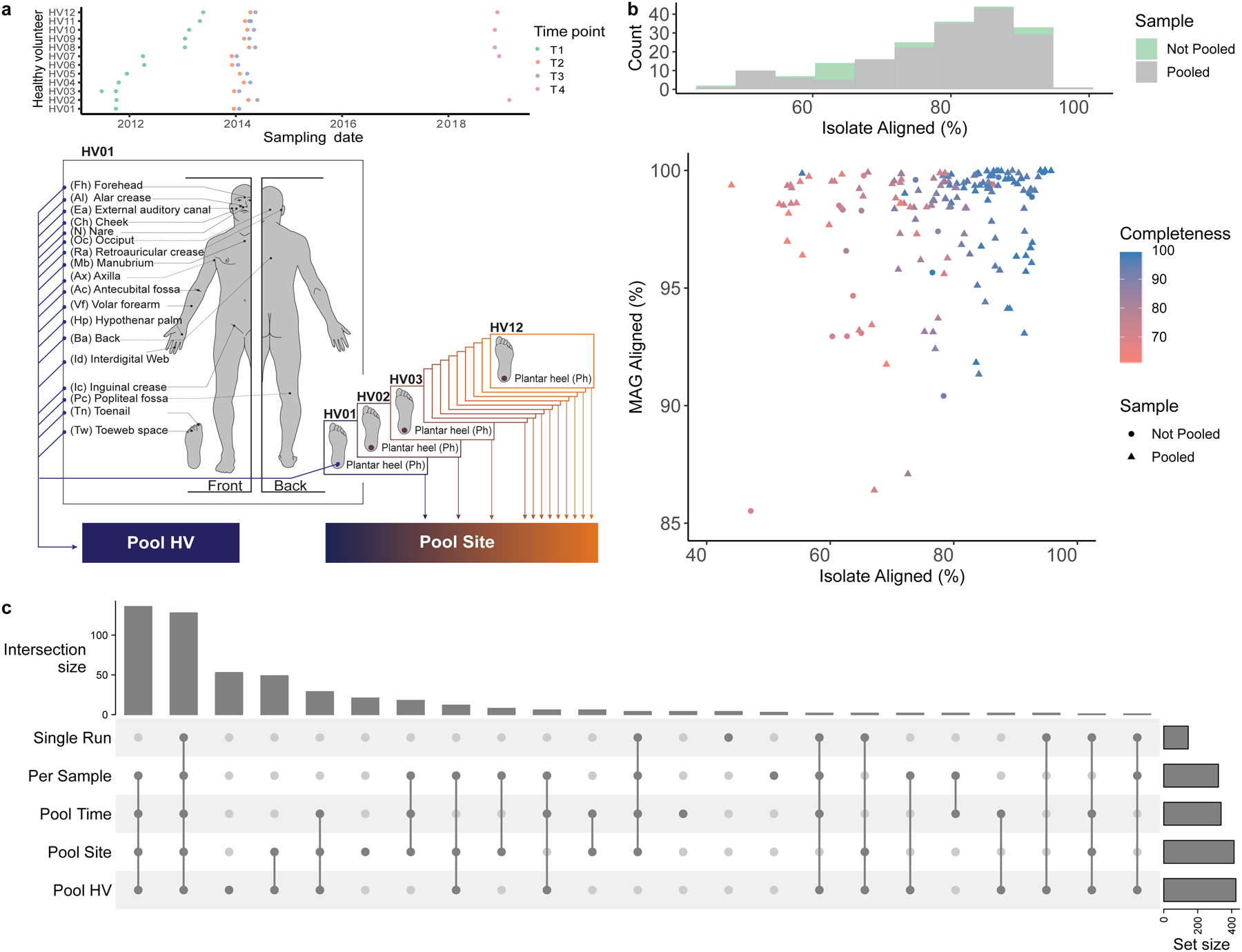
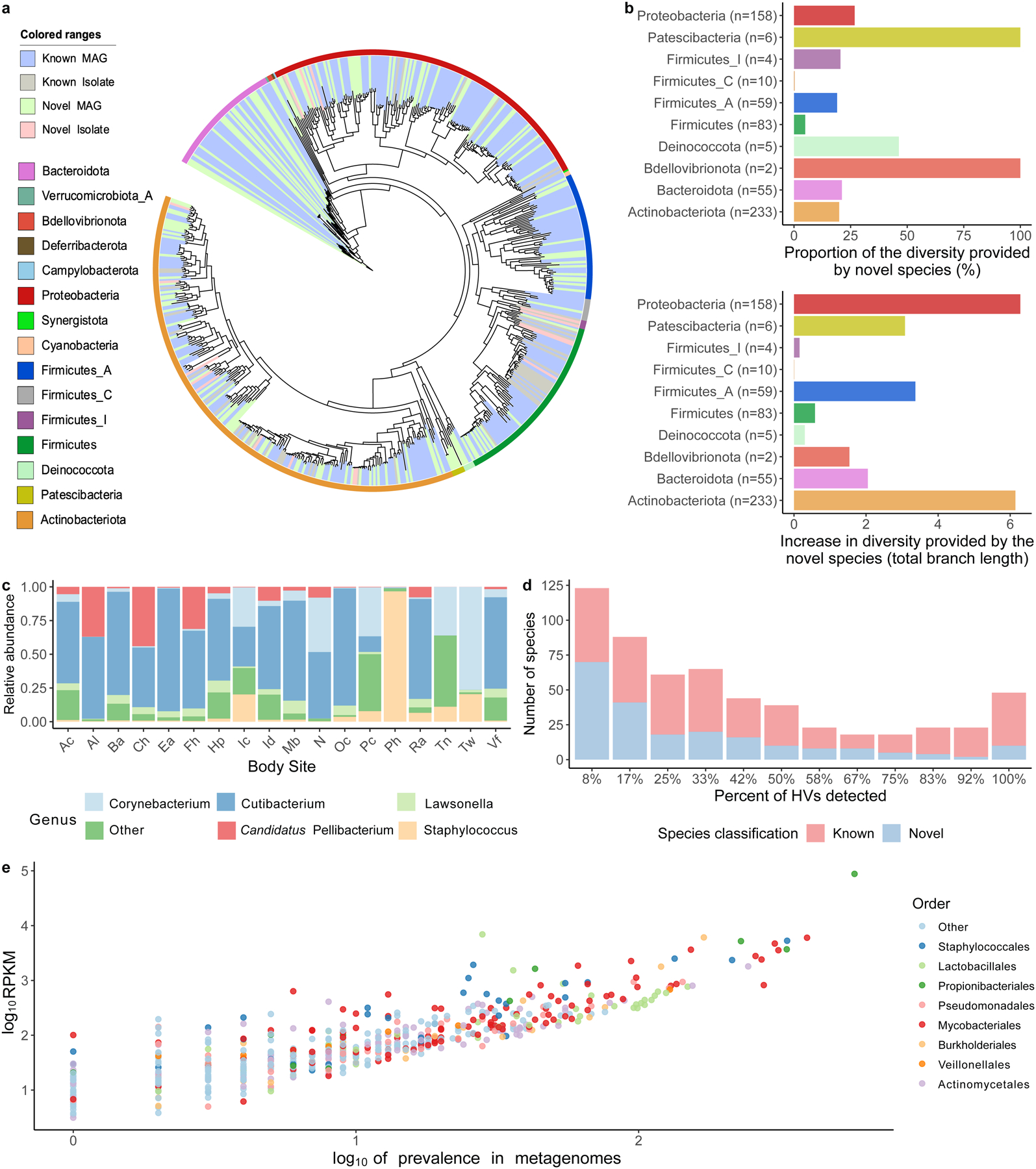
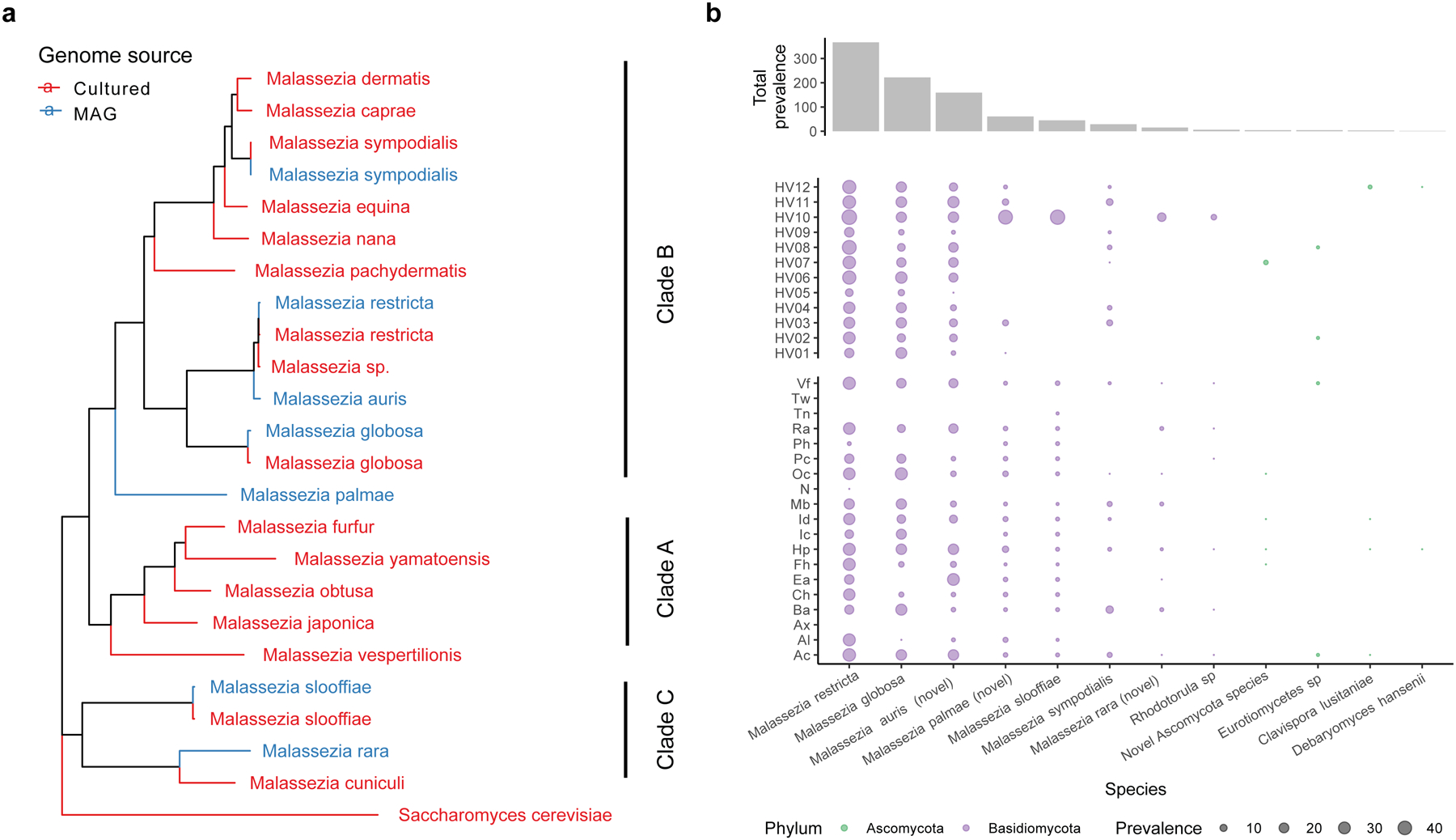
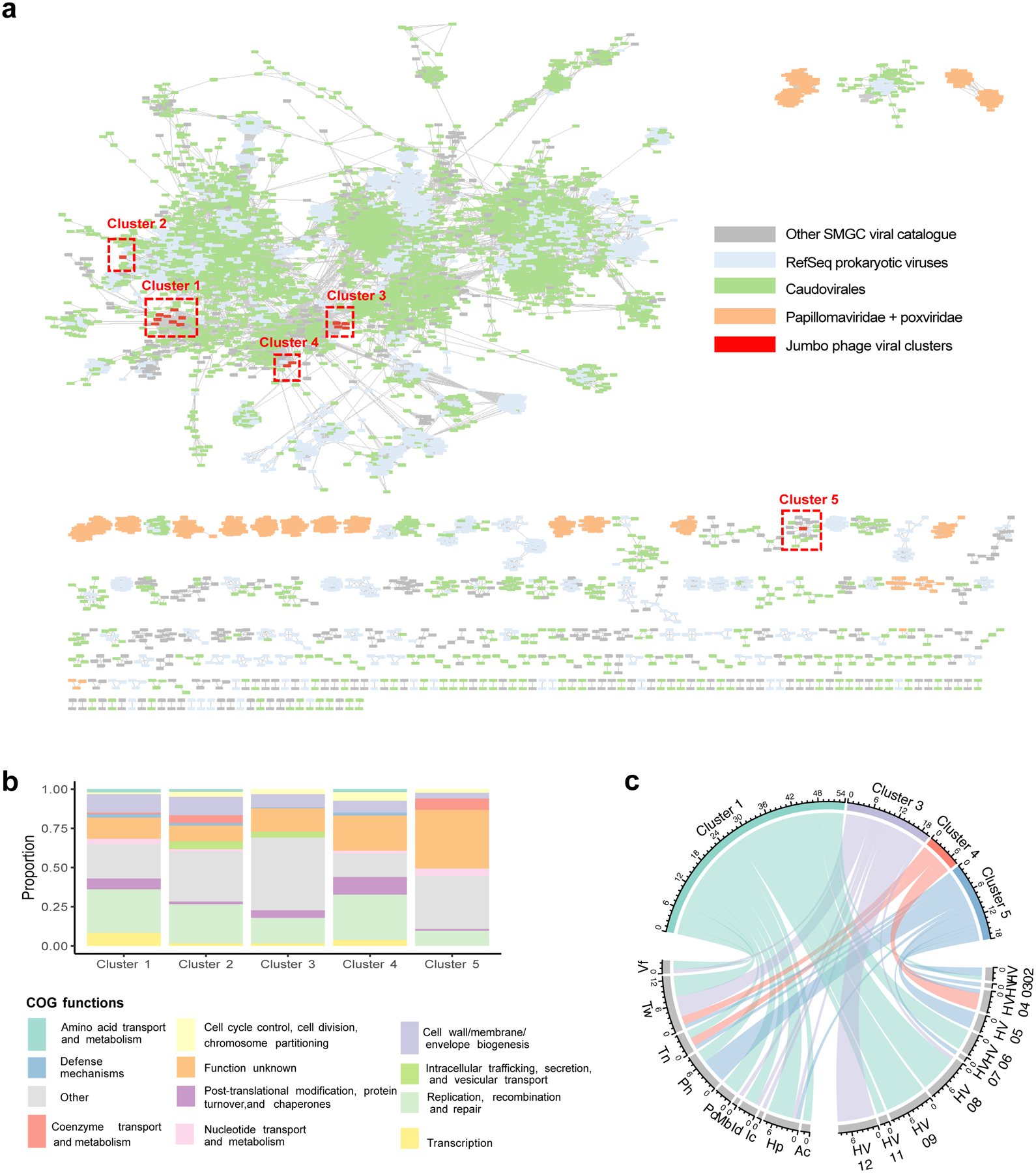
Human skin functions as a physical barrier to foreign pathogen invasion and houses numerous commensals. Shifts in the human skin microbiome have been associated with conditions ranging from acne to atopic dermatitis. Previous metagenomic investigations into the role of the skin microbiome in health or disease have found that much of the sequenced data do not match reference genomes, making it difficult to interpret metagenomic datasets. We combined bacterial cultivation and metagenomic sequencing to assemble the Skin Microbial Genome Collection (SMGC), which comprises 622 prokaryotic species derived from 7,535 metagenome-assembled genomes and 251 isolate genomes. The metagenomic datasets that we generated were combined with publicly available skin metagenomic datasets to identify members and functions of the human skin microbiome. The SMGC collection includes 174 newly identified bacterial species and 12 newly identified bacterial genera, including the abundant genus 'Candidatus Pellibacterium', which has been newly associated with the skin. The SMGC increases the characterized set of known skin bacteria by 26%. We validated the SMGC metagenome-assembled genomes by comparing them with sequenced isolates obtained from the same samples. We also recovered 12 eukaryotic species and assembled thousands of viral sequences, including newly identified clades of jumbo phages. The SMGC enables classification of a median of 85% of skin metagenomic sequences and provides a comprehensive view of skin microbiome diversity, derived primarily from samples obtained in North America.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures
References
-
- Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol 2018; 16: 143–155. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
