

Accumbal Histamine Signaling Engages Discrete Interneuron Microcircuits
- PMID: 34953589
- PMCID: PMC9012818
- DOI: 10.1016/j.biopsych.2021.10.004
Accumbal Histamine Signaling Engages Discrete Interneuron Microcircuits
Abstract
Background: Central histamine (HA) signaling modulates diverse cortical and subcortical circuits throughout the brain, including the nucleus accumbens (NAc). The NAc, a key striatal subregion directing reward-related behavior, expresses diverse HA receptor subtypes that elicit cellular and synaptic plasticity. However, the neuromodulatory capacity of HA within interneuron microcircuits in the NAc remains unknown.
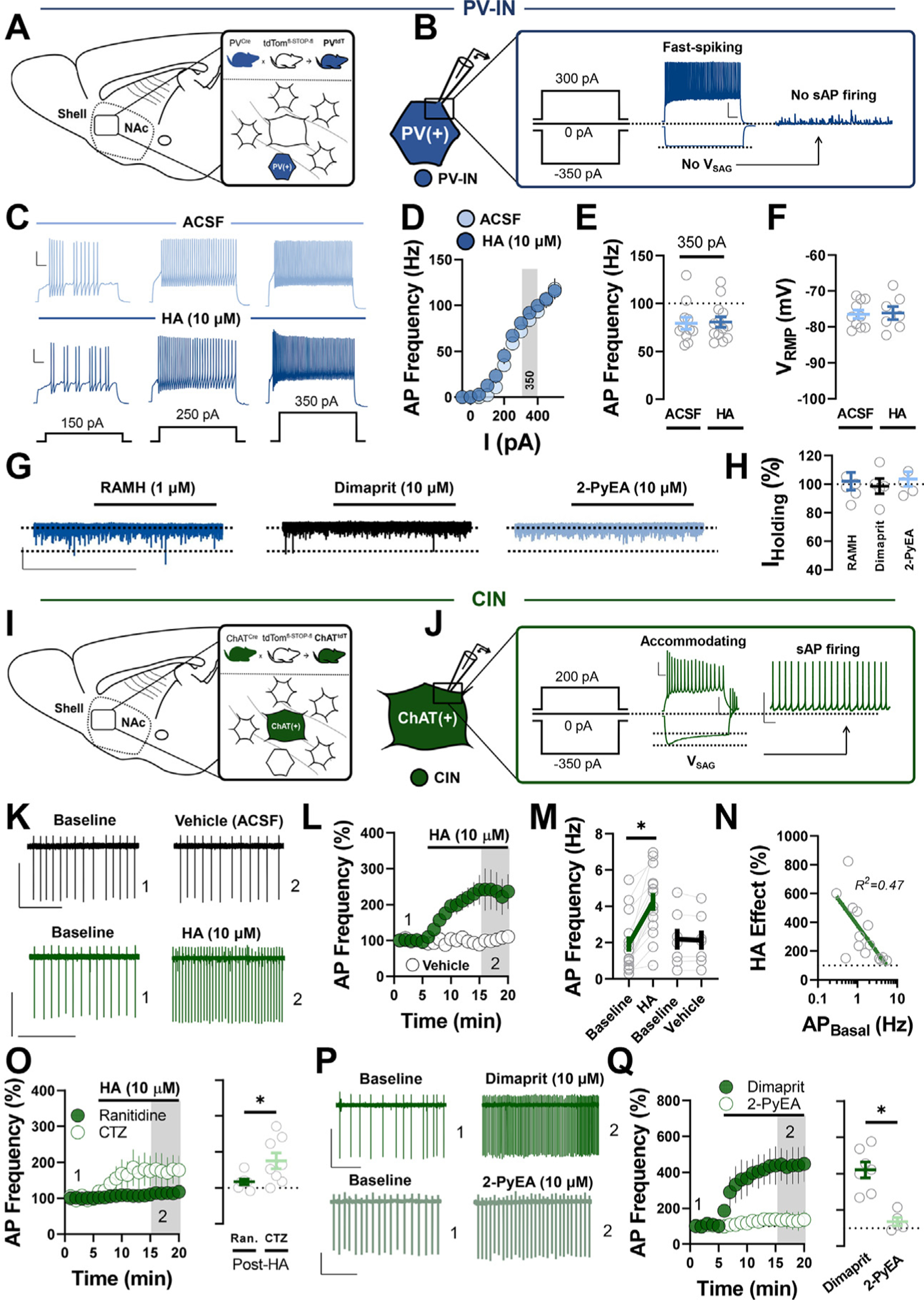
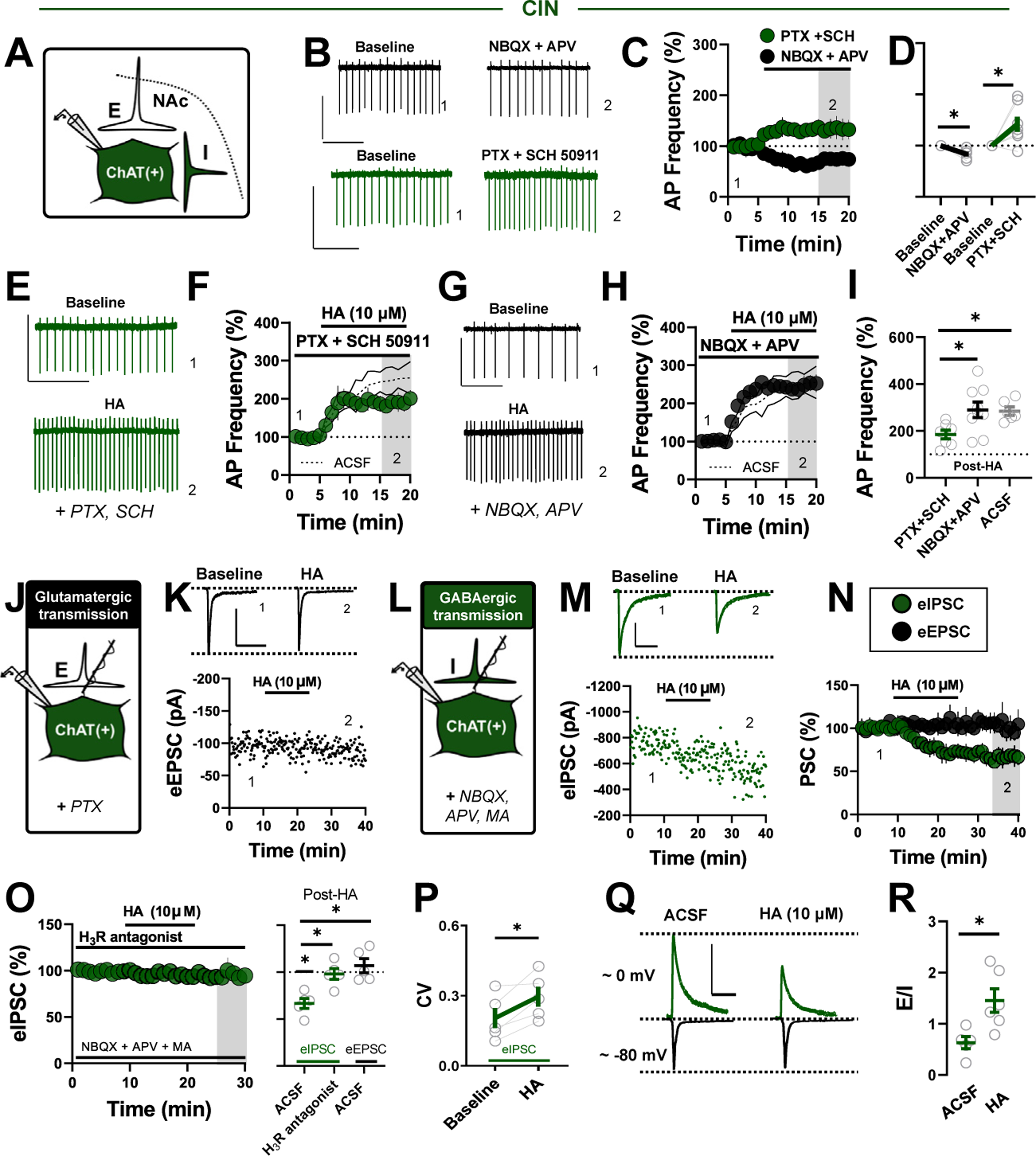
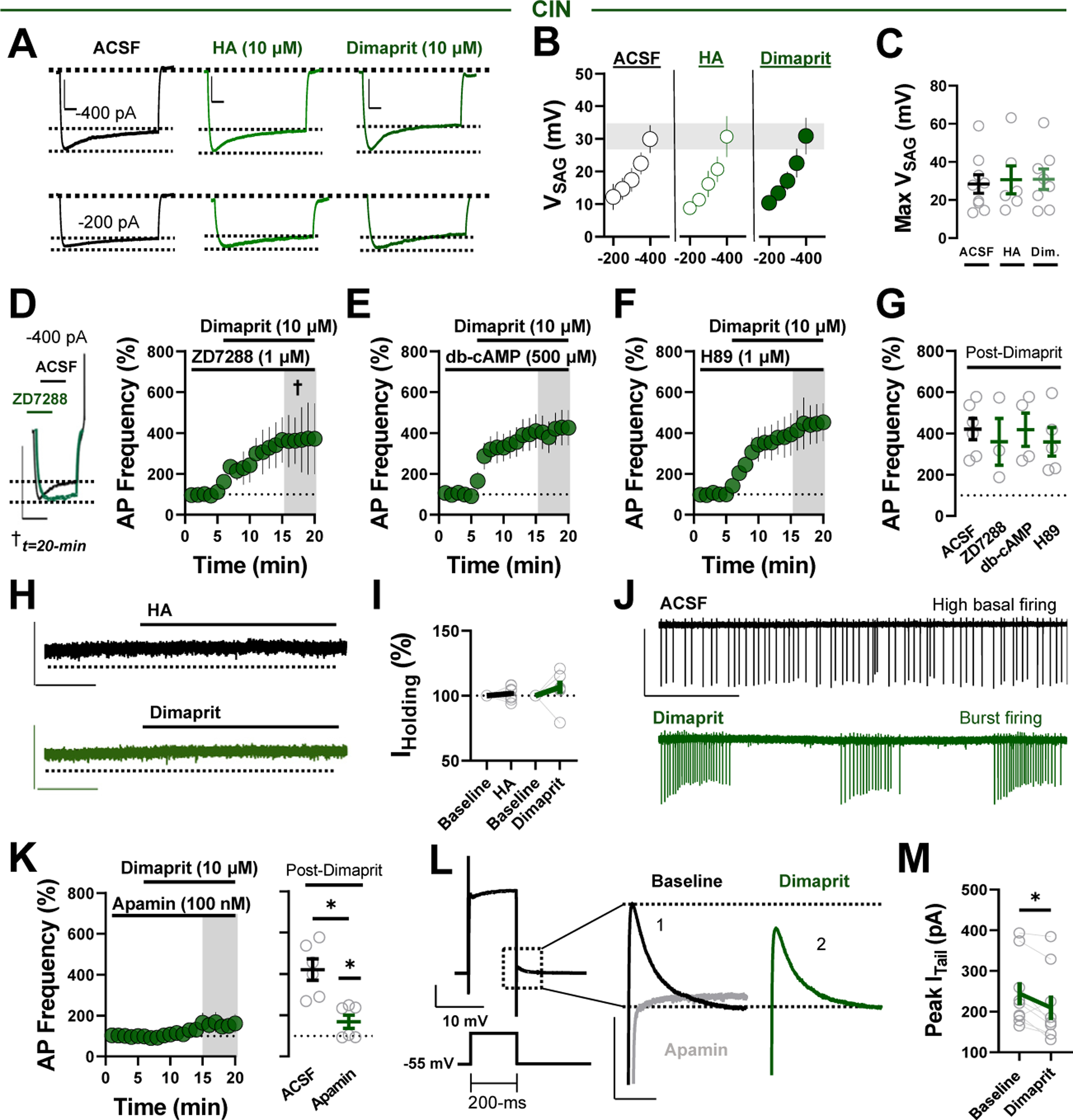
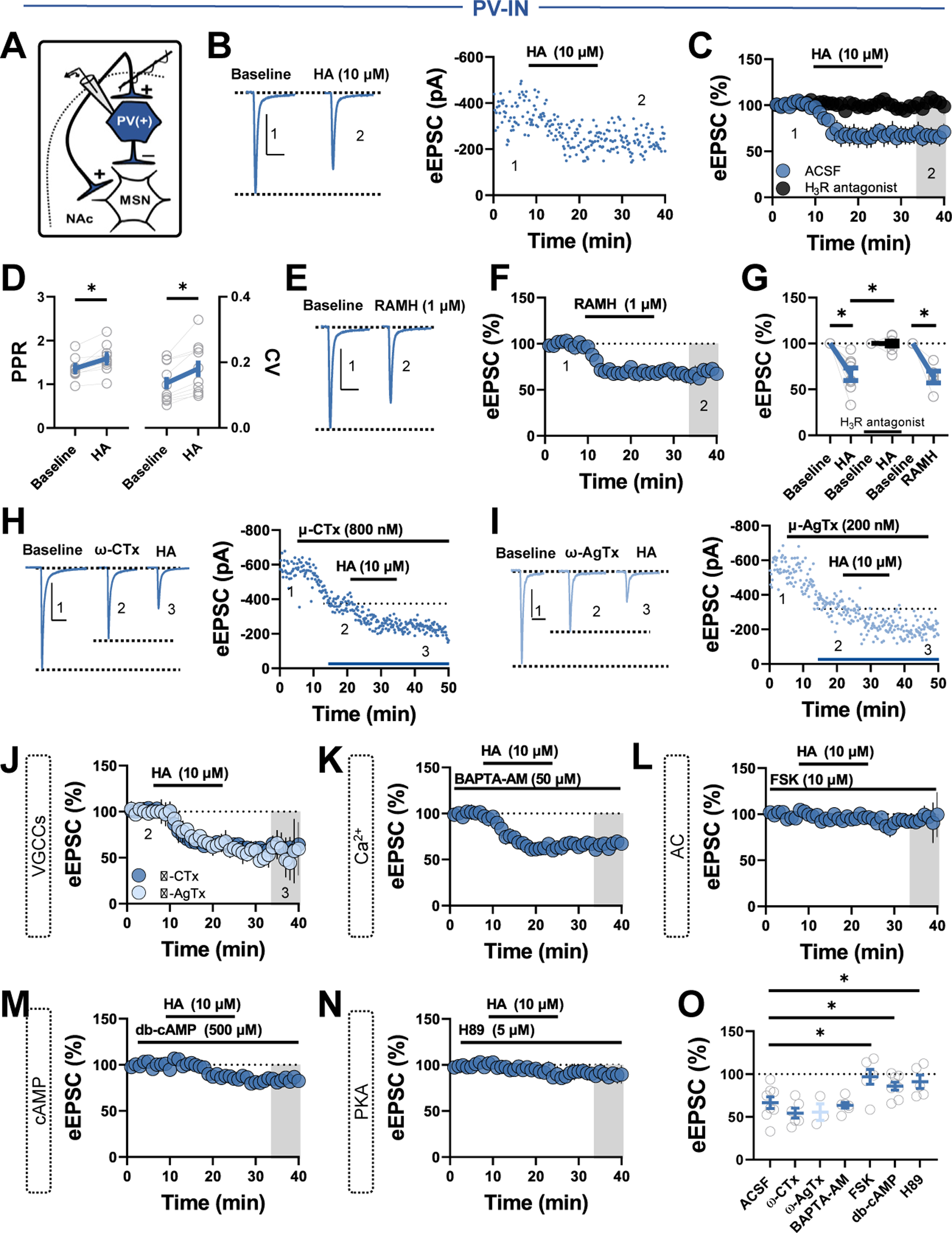
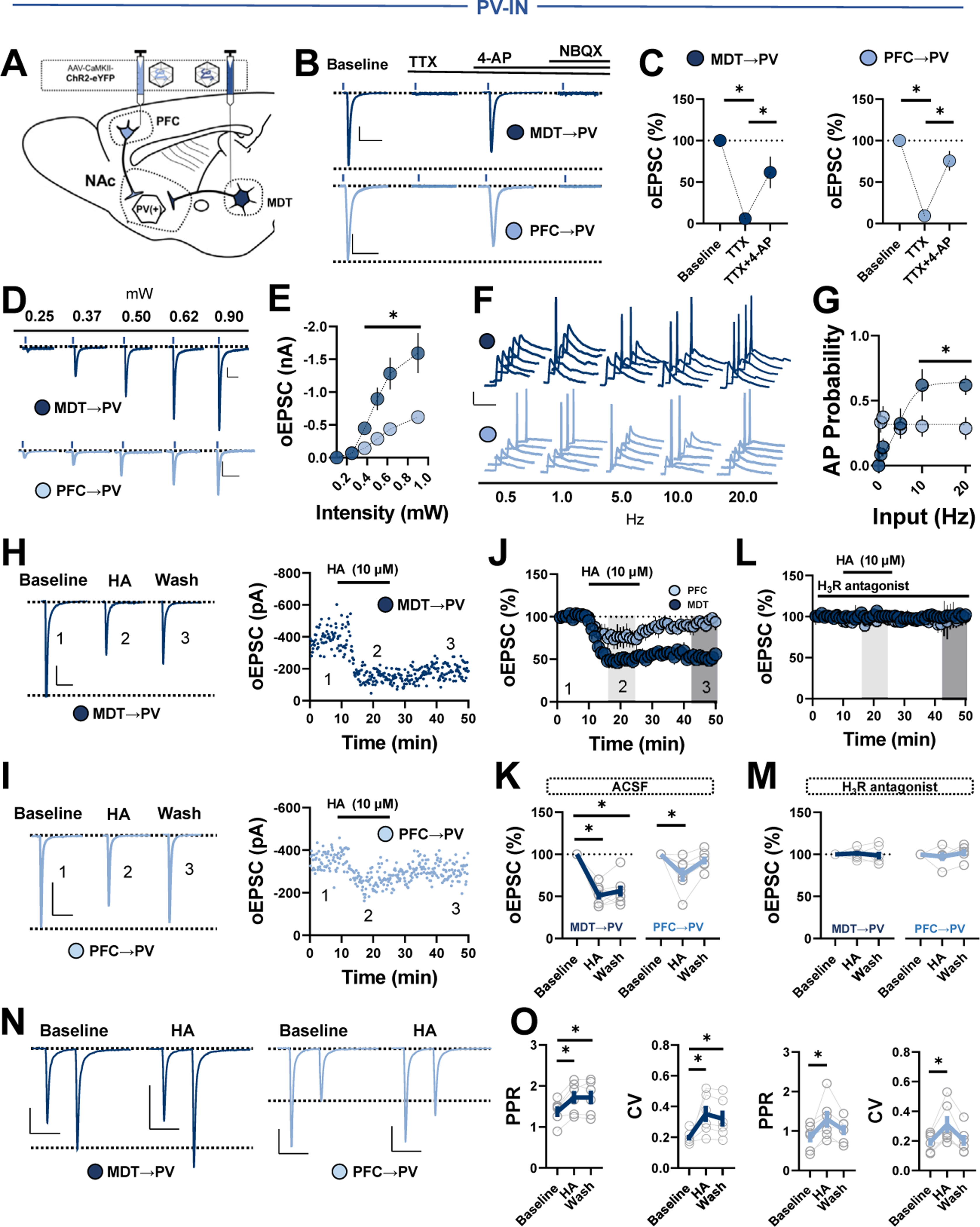
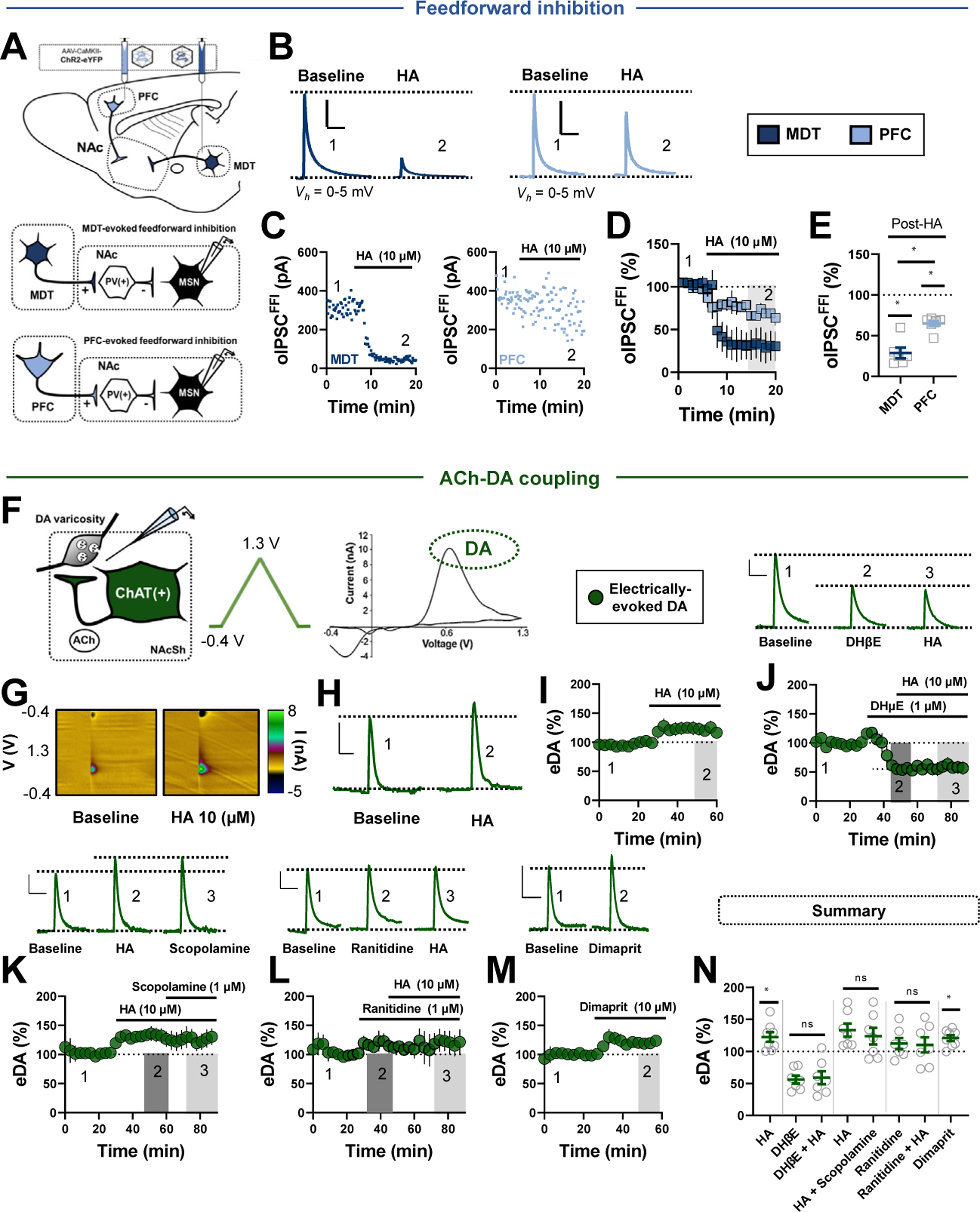
Methods: We combined electrophysiology, pharmacology, voltammetry, and optogenetics in male transgenic reporter mice to determine how HA influences microcircuit motifs controlled by parvalbumin-expressing fast-spiking interneurons (PV-INs) and tonically active cholinergic interneurons (CINs) in the NAc shell.
Results: HA enhanced CIN output through an H2 receptor (H2R)-dependent effector pathway requiring Ca2+-activated small-conductance K+ channels, with a small but discernible contribution from H1Rs and synaptic H3Rs. While PV-IN excitability was unaffected by HA, presynaptic H3Rs decreased feedforward drive onto PV-INs via AC-cAMP-PKA (adenylyl cyclase-cyclic adenosine monophosphate-protein kinase A) signaling. H3R-dependent plasticity was differentially expressed at mediodorsal thalamus and prefrontal cortex synapses onto PV-INs, with mediodorsal thalamus synapses undergoing HA-induced long-term depression. These effects triggered downstream shifts in PV-IN- and CIN-controlled microcircuits, including near-complete collapse of mediodorsal thalamus-evoked feedforward inhibition and increased mesoaccumbens dopamine release.
Conclusions: HA targets H1R, H2R, and H3Rs in the NAc shell to engage synapse- and cell type-specific mechanisms that bidirectionally regulate PV-IN and CIN microcircuit activity. These findings extend the current conceptual framework of HA signaling and offer critical insight into the modulatory potential of HA in the brain.
Keywords: Cholinergic interneurons; Dopamine; Electrophysiology; Feedforward inhibition; Histamine; Mediodorsal thalamus; Microcircuits; Nucleus accumbens; Optogenetics; Parvalbumin interneurons; Patch-clamp; Prefrontal cortex; Voltammetry.
Copyright © 2021 Society of Biological Psychiatry. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors report no biomedical financial interests or potential conflicts of interest.
Figures
Comment in
-
Histamine Regulates Accumbens Microcircuits: An Arousing Finding for Addiction Research.Biol Psychiatry. 2023 Jun 1;93(11):964-965. doi: 10.1016/j.biopsych.2023.03.008. Biol Psychiatry. 2023. PMID: 37197836 No abstract available.
References
-
- Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H (1986): Histaminergic axons in the neostriatum and cerebral cortex of the rat: A correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Res 364:114–123. - PubMed
-
- Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y, et al. (1984): Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Res 295:13–25. - PubMed
-
- Panula P, Nuutinen S (2013): The histaminergic network in the brain: 22. Basic organization and role in disease [no. 7]. Nat Rev Neurosci 14:472–487. - PubMed
-
- Passani MB, Blandina P (2011): Histamine receptors in the CNS as 23. targets for therapeutic intervention. Trends Pharmacol Sci 32:242249. - PubMed
-
- Brabant C, Alleva L, Quertemont E, Tirelli E (2010): Involvement of the 24. brain histaminergic system in addiction and addiction-related behaviors: A comprehensive review with emphasis on the potential therapeutic use of histaminergic compounds in drug dependence. Prog Neurobiol 92:421–441.. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
