

Anticancer drug resistance: An update and perspective
- PMID: 34953682
- PMCID: PMC8810687
- DOI: 10.1016/j.drup.2021.100796
Anticancer drug resistance: An update and perspective
Abstract
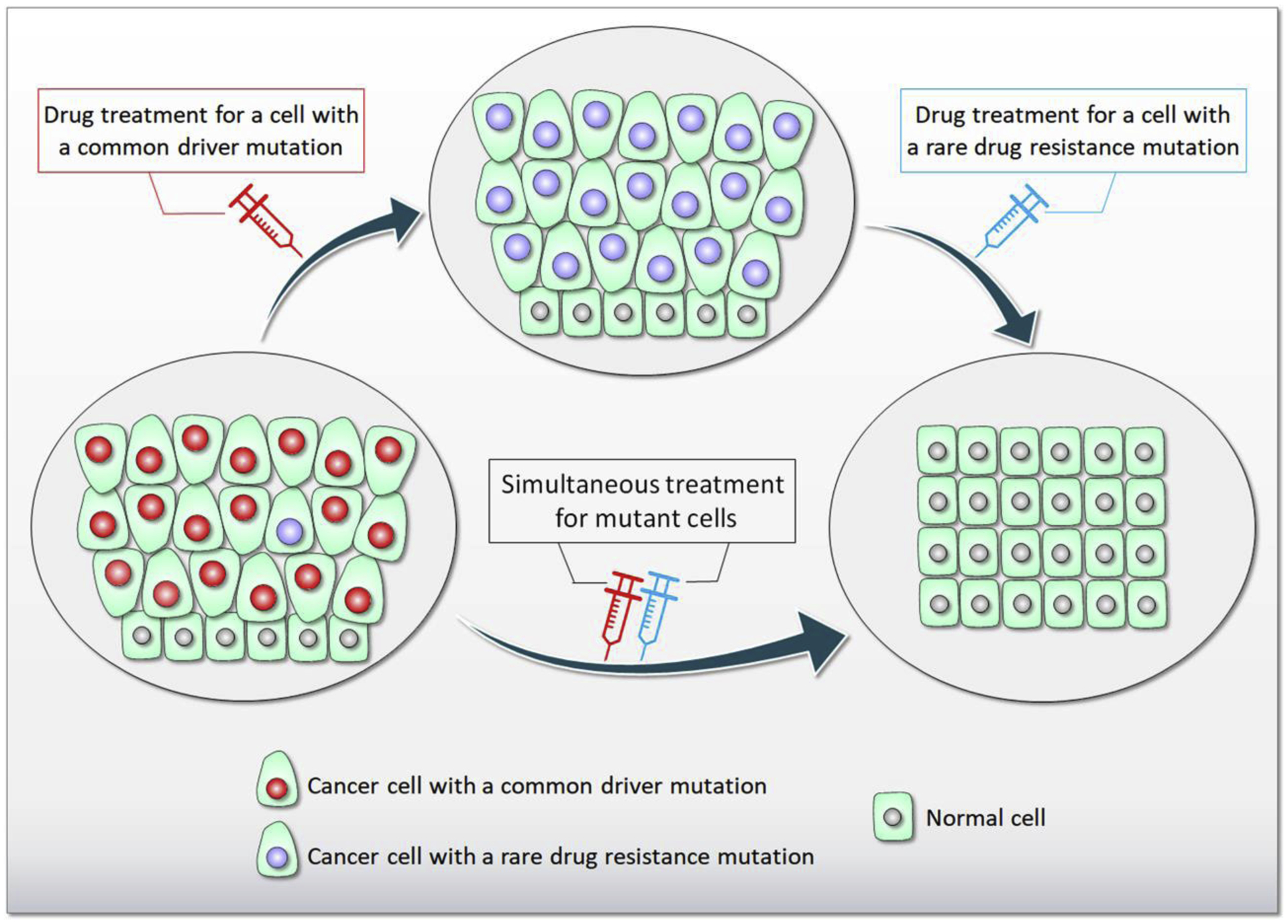
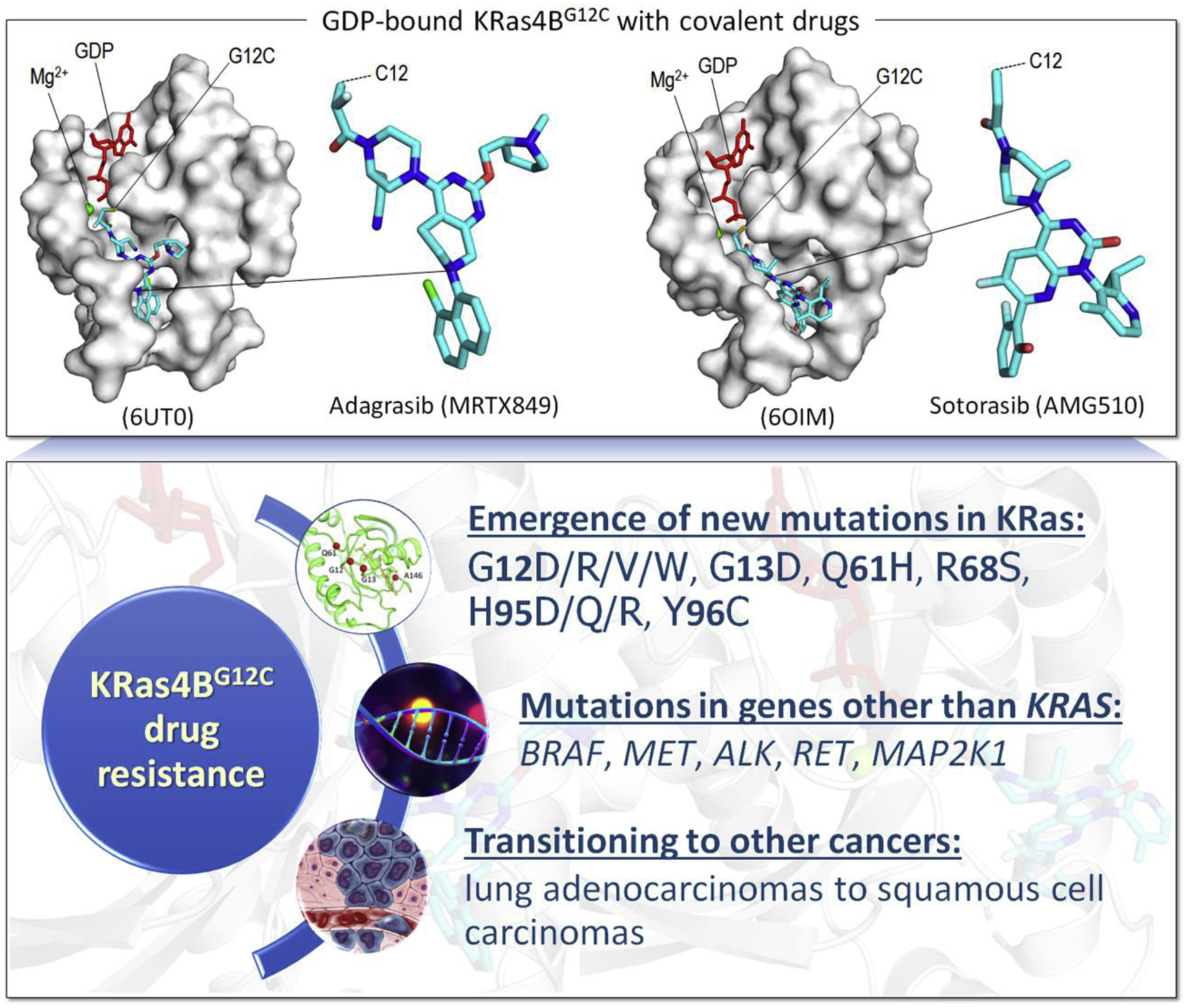
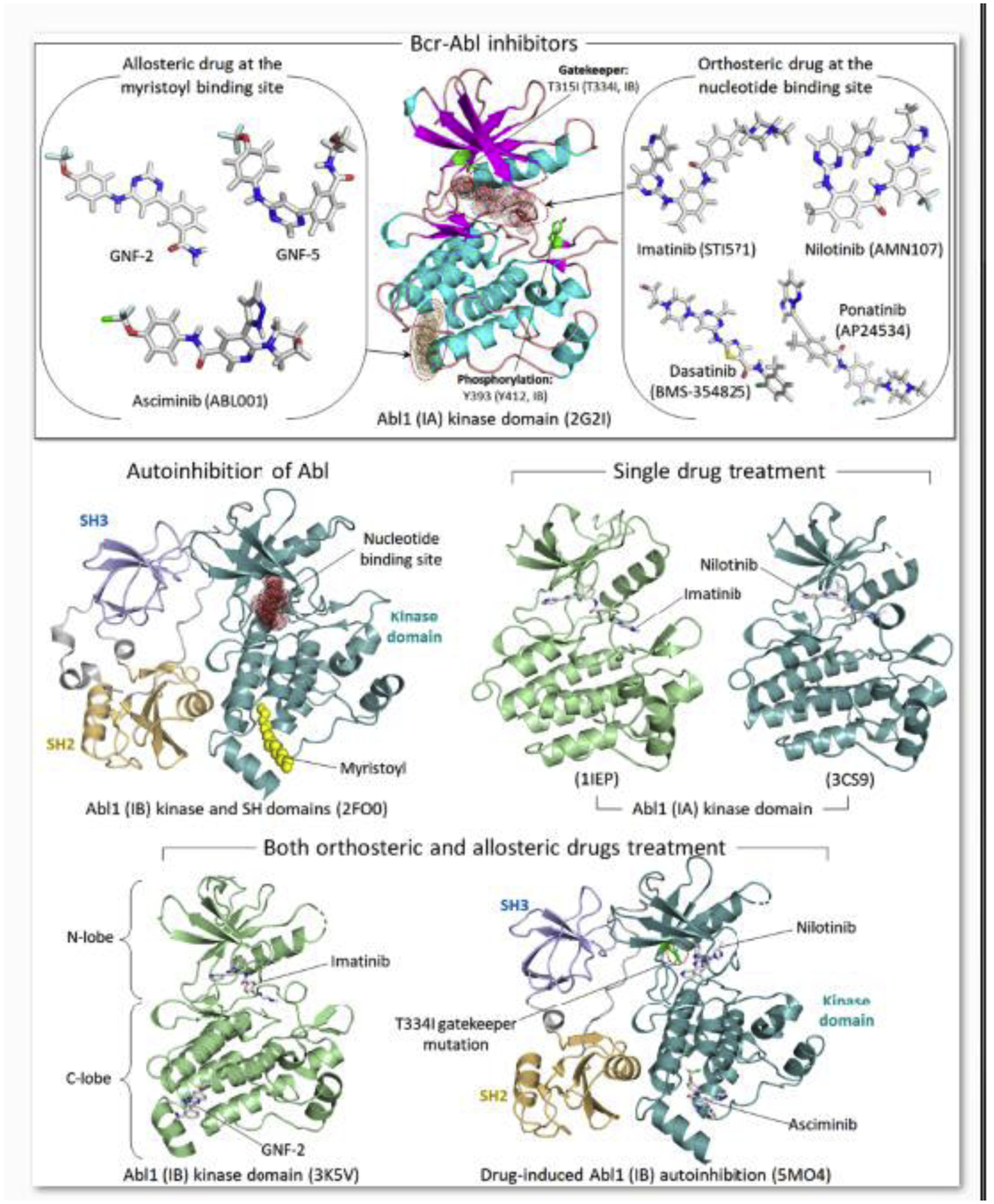
Driver mutations promote initiation and progression of cancer. Pharmacological treatment can inhibit the action of the mutant protein; however, drug resistance almost invariably emerges. Multiple studies revealed that cancer drug resistance is based upon a plethora of distinct mechanisms. Drug resistance mutations can occur in the same protein or in different proteins; as well as in the same pathway or in parallel pathways, bypassing the intercepted signaling. The dilemma that the clinical oncologist is facing is that not all the genomic alterations as well as alterations in the tumor microenvironment that facilitate cancer cell proliferation are known, and neither are the alterations that are likely to promote metastasis. For example, the common KRasG12C driver mutation emerges in different cancers. Most occur in NSCLC, but some occur, albeit to a lower extent, in colorectal cancer and pancreatic ductal carcinoma. The responses to KRasG12C inhibitors are variable and fall into three categories, (i) new point mutations in KRas, or multiple copies of KRAS G12C which lead to higher expression level of the mutant protein; (ii) mutations in genes other than KRAS; (iii) original cancer transitioning to other cancer(s). Resistance to adagrasib, an experimental antitumor agent exerting its cytotoxic effect as a covalent inhibitor of the G12C KRas, indicated that half of the cases present multiple KRas mutations as well as allele amplification. Redundant or parallel pathways included MET amplification; emerging driver mutations in NRAS, BRAF, MAP2K1, and RET; gene fusion events in ALK, RET, BRAF, RAF1, and FGFR3; and loss-of-function mutations in NF1 and PTEN tumor suppressors. In the current review we discuss the molecular mechanisms underlying drug resistance while focusing on those emerging to common targeted cancer drivers. We also address questions of why cancers with a common driver mutation are unlikely to evolve a common drug resistance mechanism, and whether one can predict the likely mechanisms that the tumor cell may develop. These vastly important and tantalizing questions in drug discovery, and broadly in precision medicine, are the focus of our present review. We end with our perspective, which calls for target combinations to be selected and prioritized with the help of the emerging massive compute power which enables artificial intelligence, and the increased gathering of data to overcome its insatiable needs.
Keywords: Cancer; Chemotherapy; Chromatin accessibility; Drug discovery; Drug resistance; Epigenetics; Interactome; MAPK; Precision medicine; Single cell; Transcriptomics.
Copyright © 2021. Published by Elsevier Ltd.
Conflict of interest statement
Declaration of Competing Interest
The authors report no declarations of interest.
Figures
References
-
- Adrian FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, Mestan J, Fabbro D, Gray NS, 2006. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol 2, 95–102. - PubMed
-
- Aissa AF, Islam A, Ariss MM, Go CC, Rader AE, Conrardy RD, Gajda AM, Rubio-Perez C, Valyi-Nagy K, Pasquinelli M, Feldman LE, Green SJ, Lopez-Bigas N, Frolov MV, Benevolenskaya EV, 2021. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat Commun 12, 1628. - PMC - PubMed
-
- Akdemir KC, Le VT, Kim JM, Killcoyne S, King DA, Lin YP, Tian Y, Inoue A, Amin SB, Robinson FS, Nimmakayalu M, Herrera RE, Lynn EJ, Chan K, Seth S, Klimczak LJ, Gerstung M, Gordenin DA, O’Brien J, Li L, Deribe YL, Verhaak RG, Campbell PJ, Fitzgerald R, Morrison AJ, Dixon JR, Andrew Futreal P, 2020b. Somatic mutation distributions in cancer genomes vary with three-dimensional chromatin structure. Nat Genet 52, 1178–1188. - PMC - PubMed
-
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, Cancer as a Microevolutionary Process. Molecular Biology, Garland Science, New York: (2002).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
