

Variants in Mitochondrial ATP Synthase Cause Variable Neurologic Phenotypes
- PMID: 34954817
- PMCID: PMC9939050
- DOI: 10.1002/ana.26293
Variants in Mitochondrial ATP Synthase Cause Variable Neurologic Phenotypes
Abstract
Objective: ATP synthase (ATPase) is responsible for the majority of ATP production. Nevertheless, disease phenotypes associated with mutations in ATPase subunits are extremely rare. We aimed at expanding the spectrum of ATPase-related diseases.
Methods: Whole-exome sequencing in cohorts with 2,962 patients diagnosed with mitochondrial disease and/or dystonia and international collaboration were used to identify deleterious variants in ATPase-encoding genes. Findings were complemented by transcriptional and proteomic profiling of patient fibroblasts. ATPase integrity and activity were assayed using cells and tissues from 5 patients.
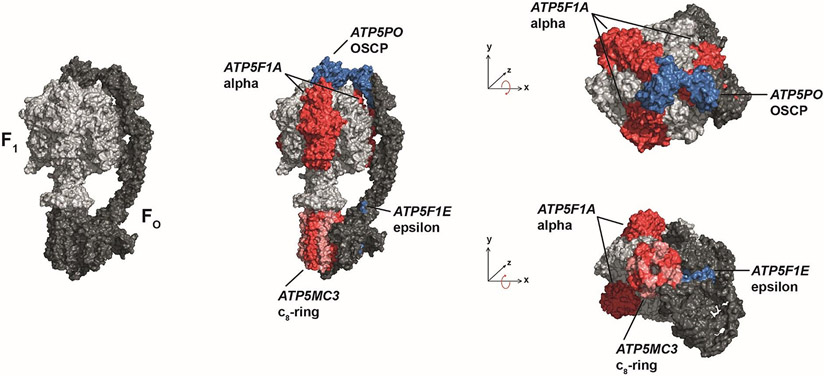
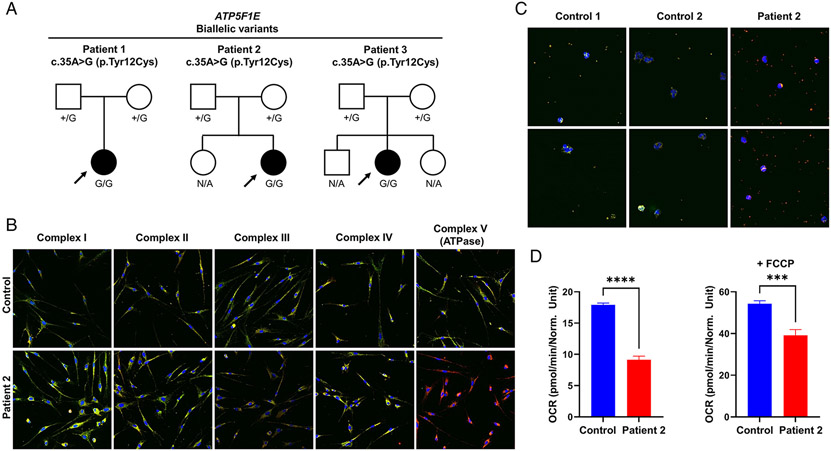
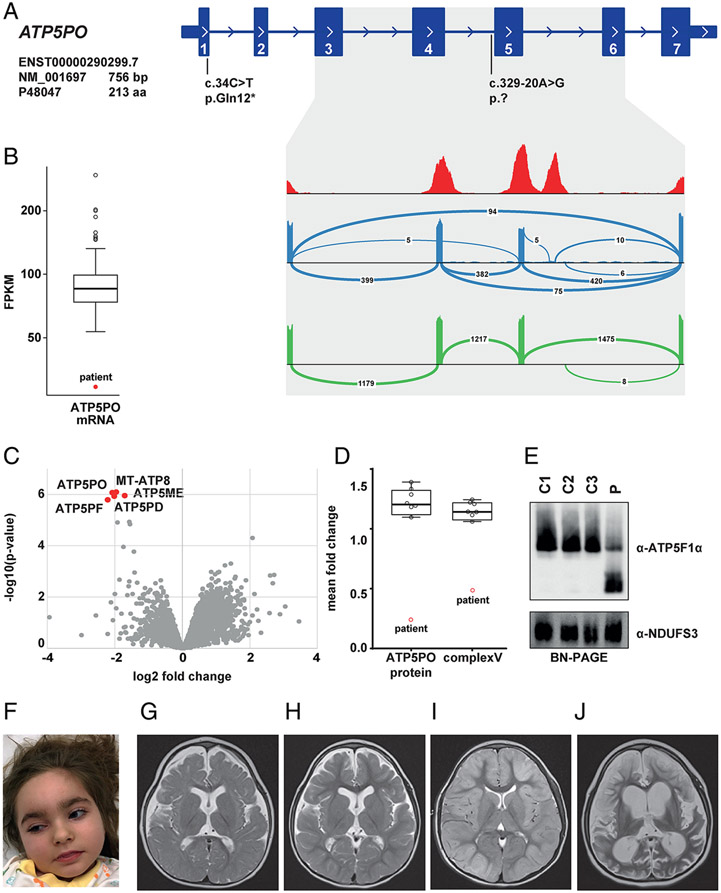
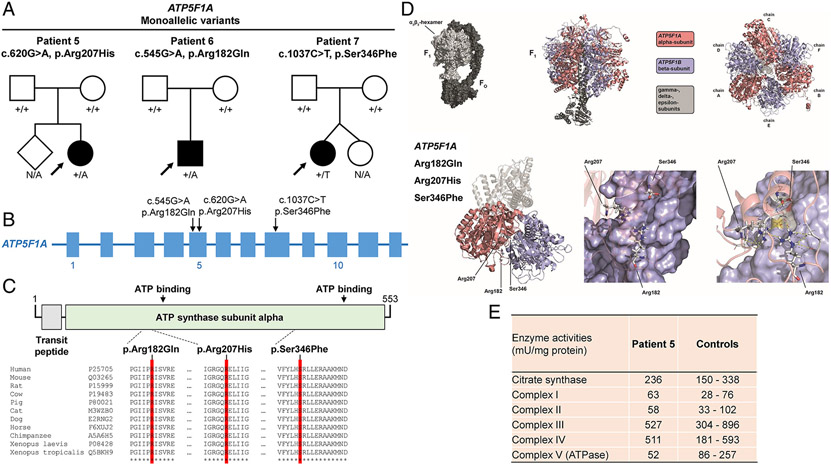
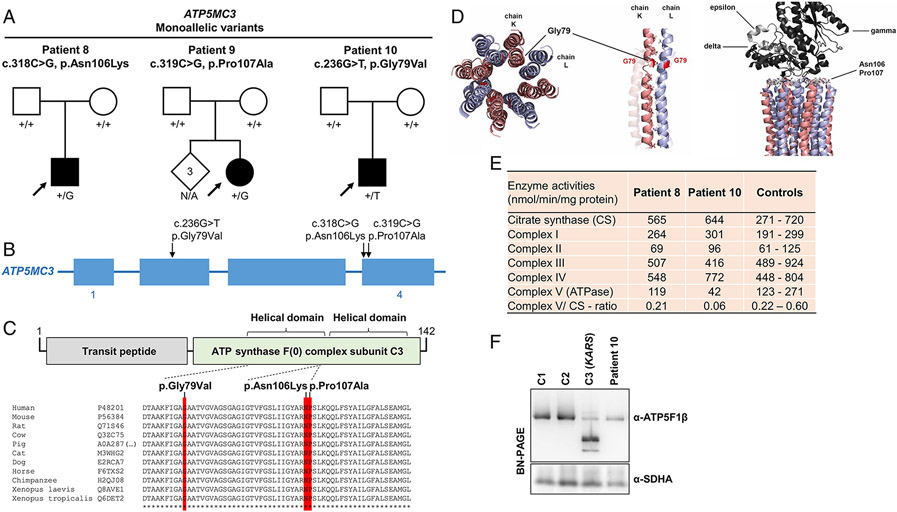
Results: We present 10 total individuals with biallelic or de novo monoallelic variants in nuclear ATPase subunit genes. Three unrelated patients showed the same homozygous missense ATP5F1E mutation (including one published case). An intronic splice-disrupting alteration in compound heterozygosity with a nonsense variant in ATP5PO was found in one patient. Three patients had de novo heterozygous missense variants in ATP5F1A, whereas another 3 were heterozygous for ATP5MC3 de novo missense changes. Bioinformatics methods and populational data supported the variants' pathogenicity. Immunohistochemistry, proteomics, and/or immunoblotting revealed significantly reduced ATPase amounts in association to ATP5F1E and ATP5PO mutations. Diminished activity and/or defective assembly of ATPase was demonstrated by enzymatic assays and/or immunoblotting in patient samples bearing ATP5F1A-p.Arg207His, ATP5MC3-p.Gly79Val, and ATP5MC3-p.Asn106Lys. The associated clinical profiles were heterogeneous, ranging from hypotonia with spontaneous resolution (1/10) to epilepsy with early death (1/10) or variable persistent abnormalities, including movement disorders, developmental delay, intellectual disability, hyperlactatemia, and other neurologic and systemic features. Although potentially reflecting an ascertainment bias, dystonia was common (7/10).
Interpretation: Our results establish evidence for a previously unrecognized role of ATPase nuclear-gene defects in phenotypes characterized by neurodevelopmental and neurodegenerative features. ANN NEUROL 2022;91:225-237.
© 2021 The Authors. Annals of Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
Potential Conflicts of Interest
None of the authors has any relevant conflict of interest to declare.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- Else Kröner-Fresenius-Stiftung
- R01 HG009141/HG/NHGRI NIH HHS/United States
- 01GM1906A/Federal Ministry of Education and Research
- Technische Universität München
- R00 HL143036/HL/NHLBI NIH HHS/United States
- U01 HG011755/HG/NHGRI NIH HHS/United States
- I4704-B/Federal Ministry of Education and Research
- 01KU2016A/Federal Ministry of Education and Research
- I 4695/FWF_/Austrian Science Fund FWF/Austria
- R01 NS106298/NS/NINDS NIH HHS/United States
- Medizinische Universität Innsbruck
- NV19-04-00233/Ministry of Education
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- 01GM1920A/Federal Ministry of Education and Research
- Research Foundation
- Charles University
- I4695-B/Federal Ministry of Education and Research
- Helmholtz Zentrum München
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
