

Osteosarcoma and Metastasis
- PMID: 34956899
- PMCID: PMC8702962
- DOI: 10.3389/fonc.2021.780264
Osteosarcoma and Metastasis
Abstract
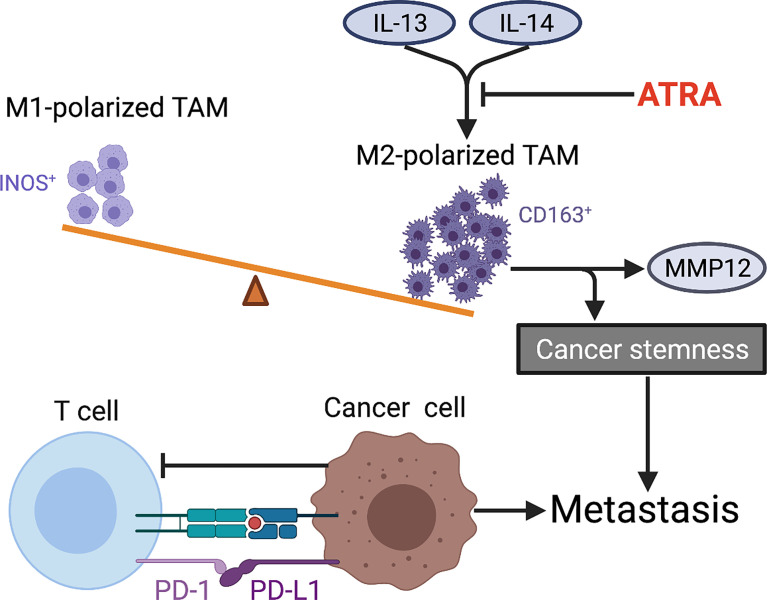
Osteosarcoma is the most common primary bone malignancy in adolescents. Its high propensity to metastasize is the leading cause for treatment failure and poor prognosis. Although the research of osteosarcoma has greatly expanded in the past decades, the knowledge and new therapy strategies targeting metastatic progression remain sparse. The prognosis of patients with metastasis is still unsatisfactory. There is resonating urgency for a thorough and deeper understanding of molecular mechanisms underlying osteosarcoma to develop innovative therapies targeting metastasis. Toward the goal of elaborating the characteristics and biological behavior of metastatic osteosarcoma, it is essential to combine the diverse investigations that are performed at molecular, cellular, and animal levels from basic research to clinical translation spanning chemical, physical sciences, and biology. This review focuses on the metastatic process, regulatory networks involving key molecules and signaling pathways, the role of microenvironment, osteoclast, angiogenesis, metabolism, immunity, and noncoding RNAs in osteosarcoma metastasis. The aim of this review is to provide an overview of current research advances, with the hope to discovery druggable targets and promising therapy strategies for osteosarcoma metastasis and thus to overcome this clinical impasse.
Keywords: metabolism; metastasis; microenvironment; noncoding RNAs; osteosarcoma.
Copyright © 2021 Sheng, Gao, Yang and Wu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
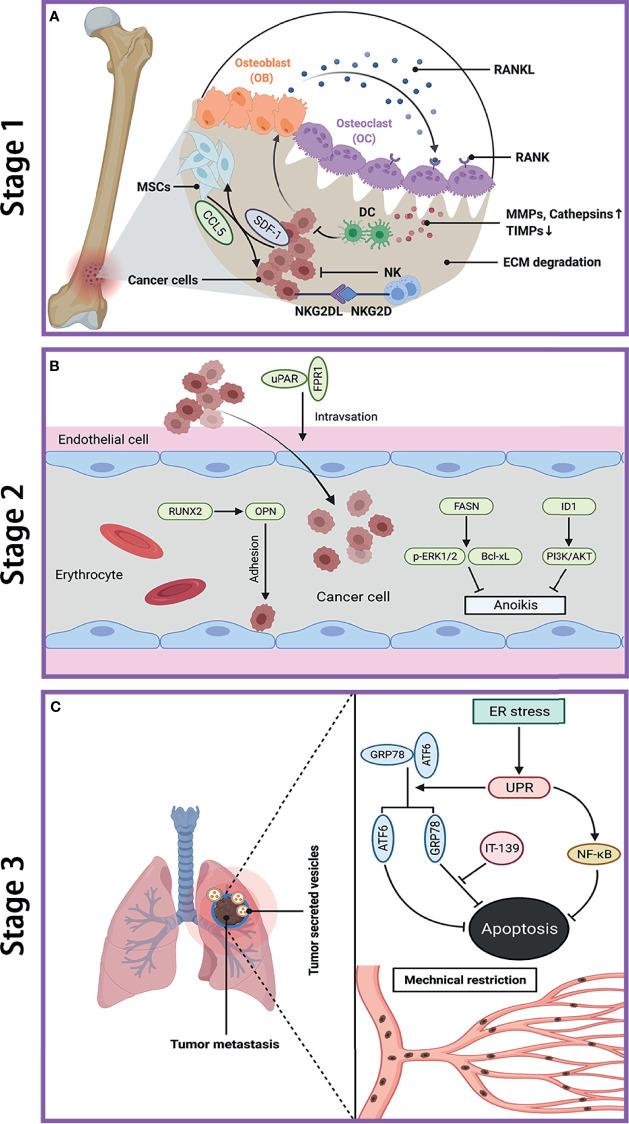
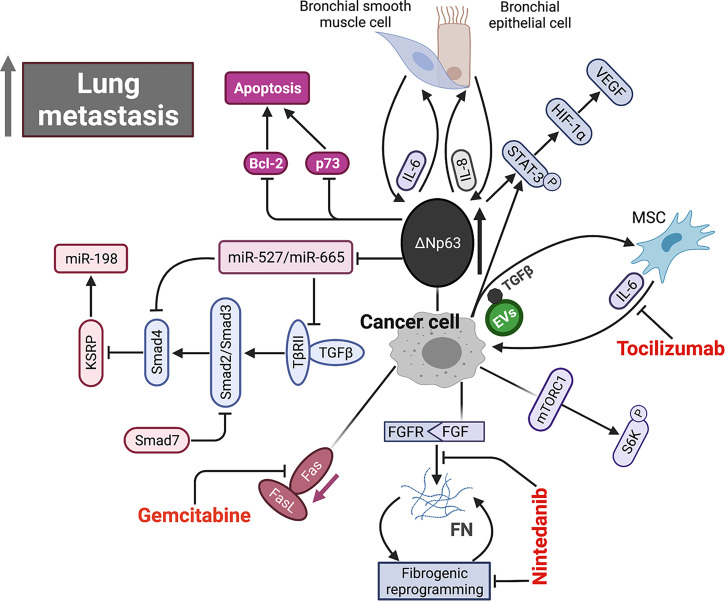
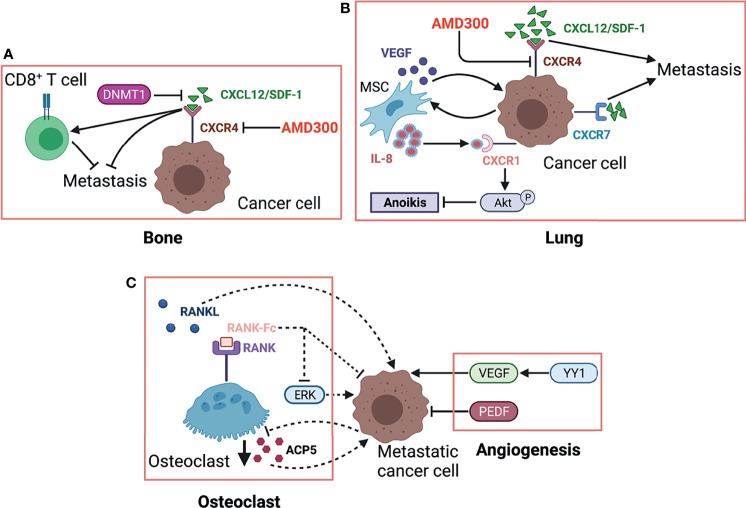
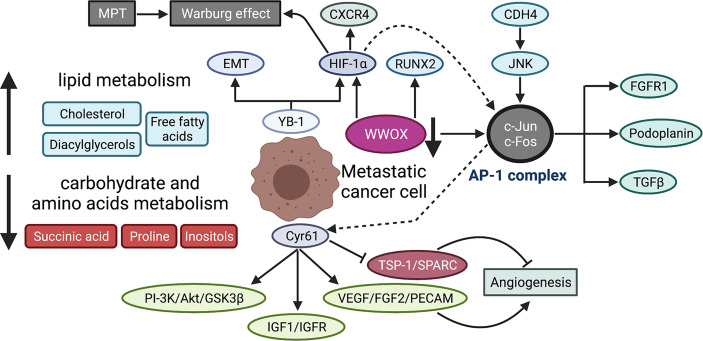
References
-
- Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, et al. Prognostic Factors in High-Grade Osteosarcoma of the Extremities or Trunk: An Analysis of 1,702 Patients Treated on Neoadjuvant Cooperative Osteosarcoma Study Group Protocols. J Clin Oncol (2002) 20(3):776–90. doi: 10.1200/JCO.2002.20.3.776 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous