

Structural mechanism for the selective phosphorylation of DNA-loaded MCM double hexamers by the Dbf4-dependent kinase
- PMID: 34963704
- PMCID: PMC8770131
- DOI: 10.1038/s41594-021-00698-z
Structural mechanism for the selective phosphorylation of DNA-loaded MCM double hexamers by the Dbf4-dependent kinase
Abstract
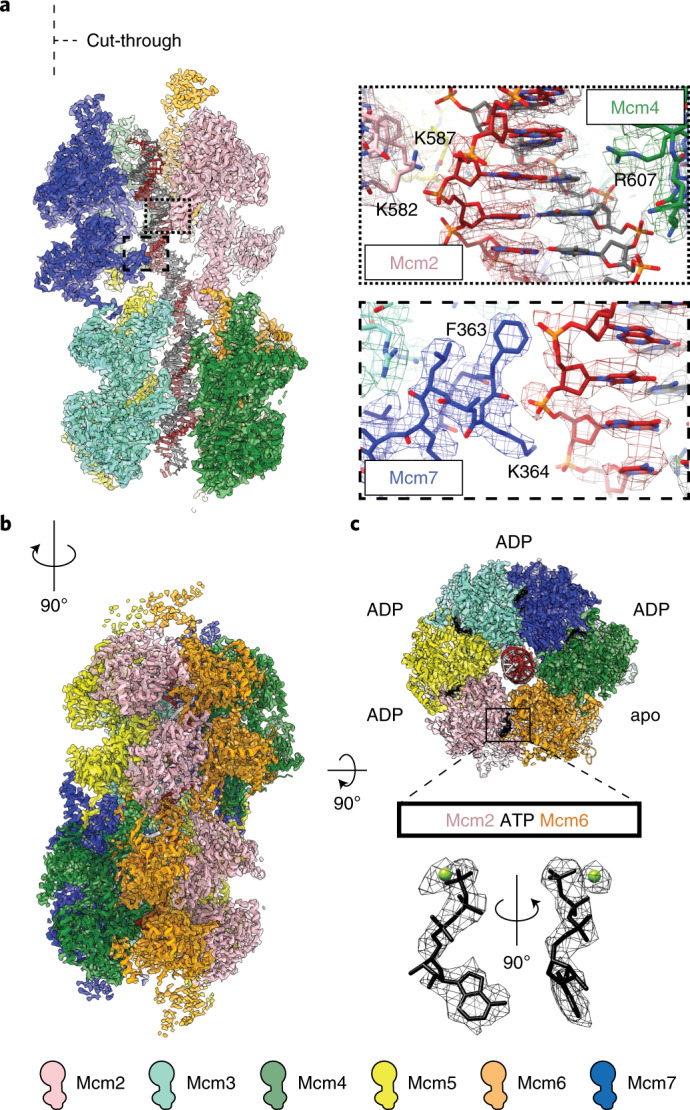
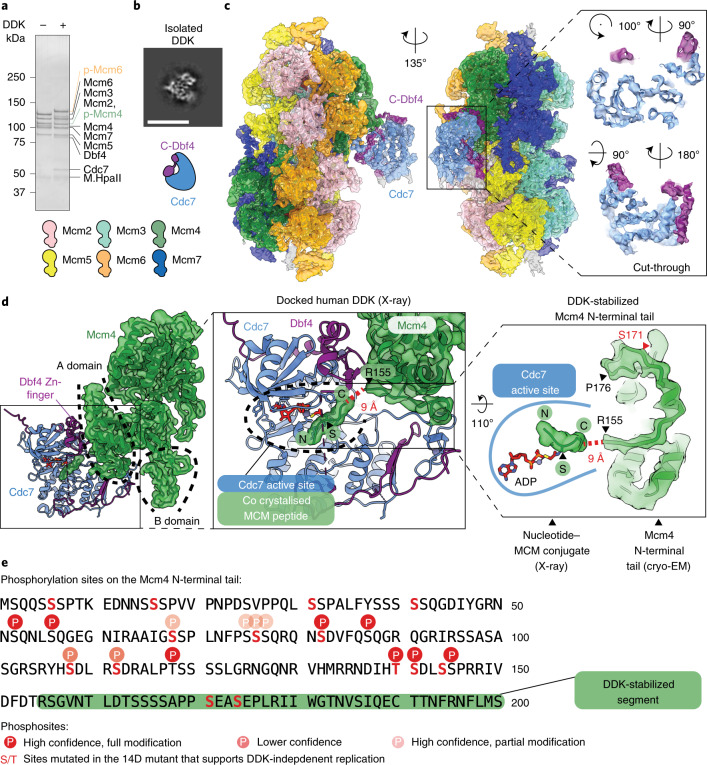
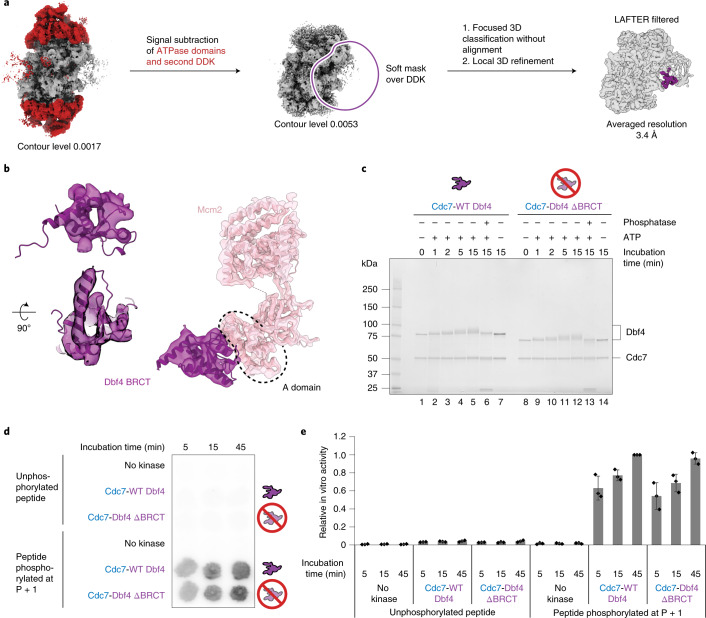
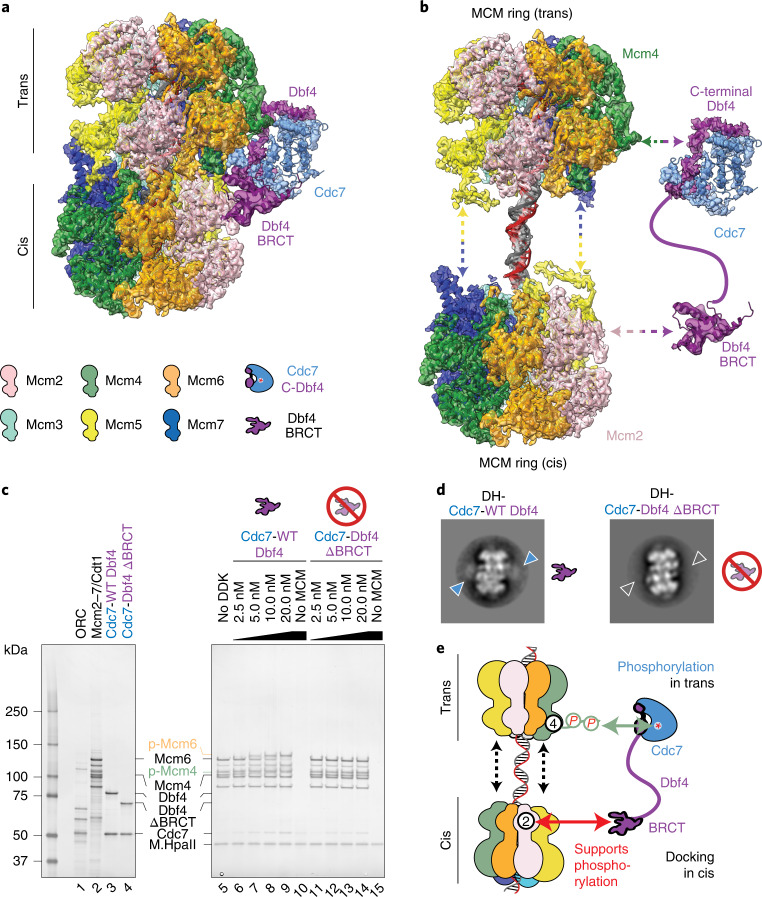
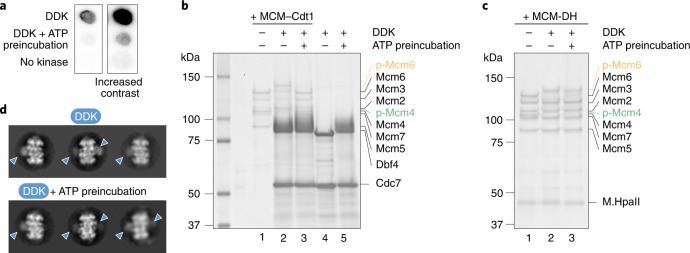
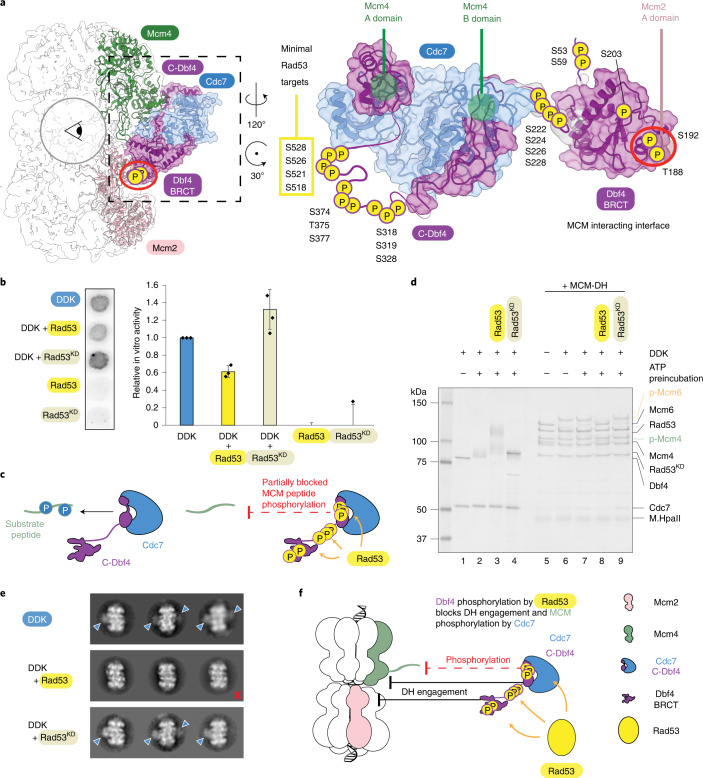
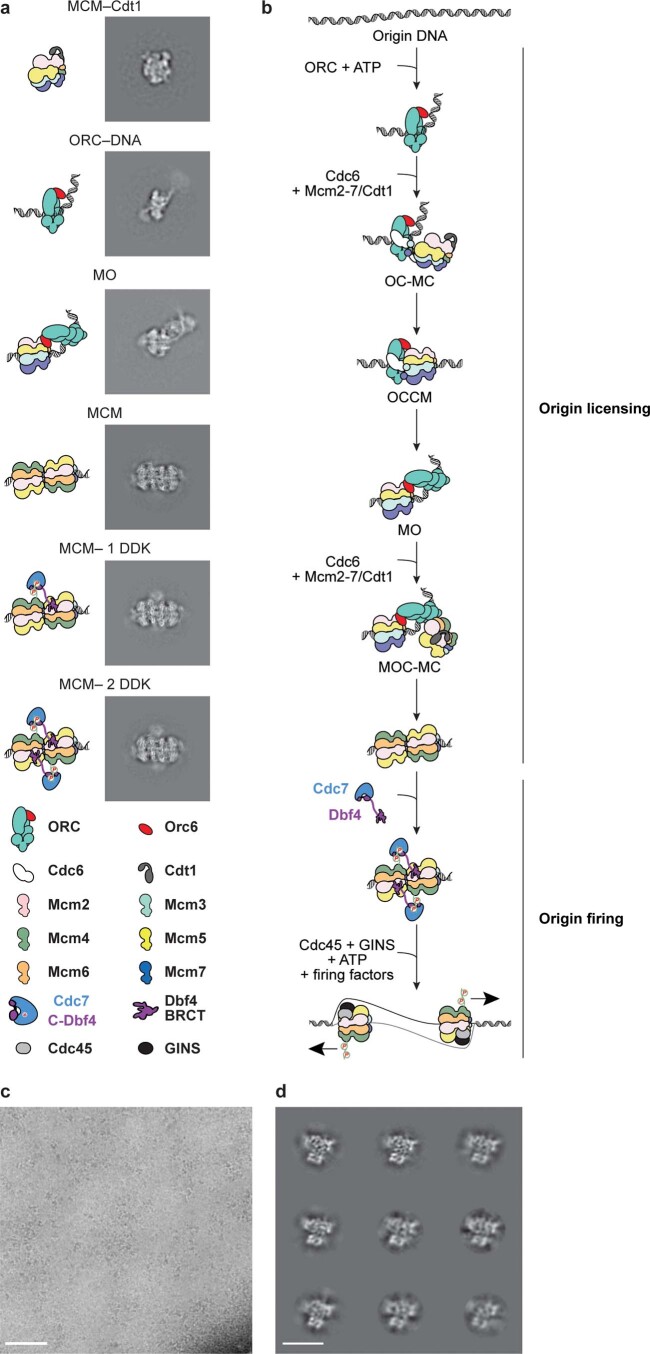
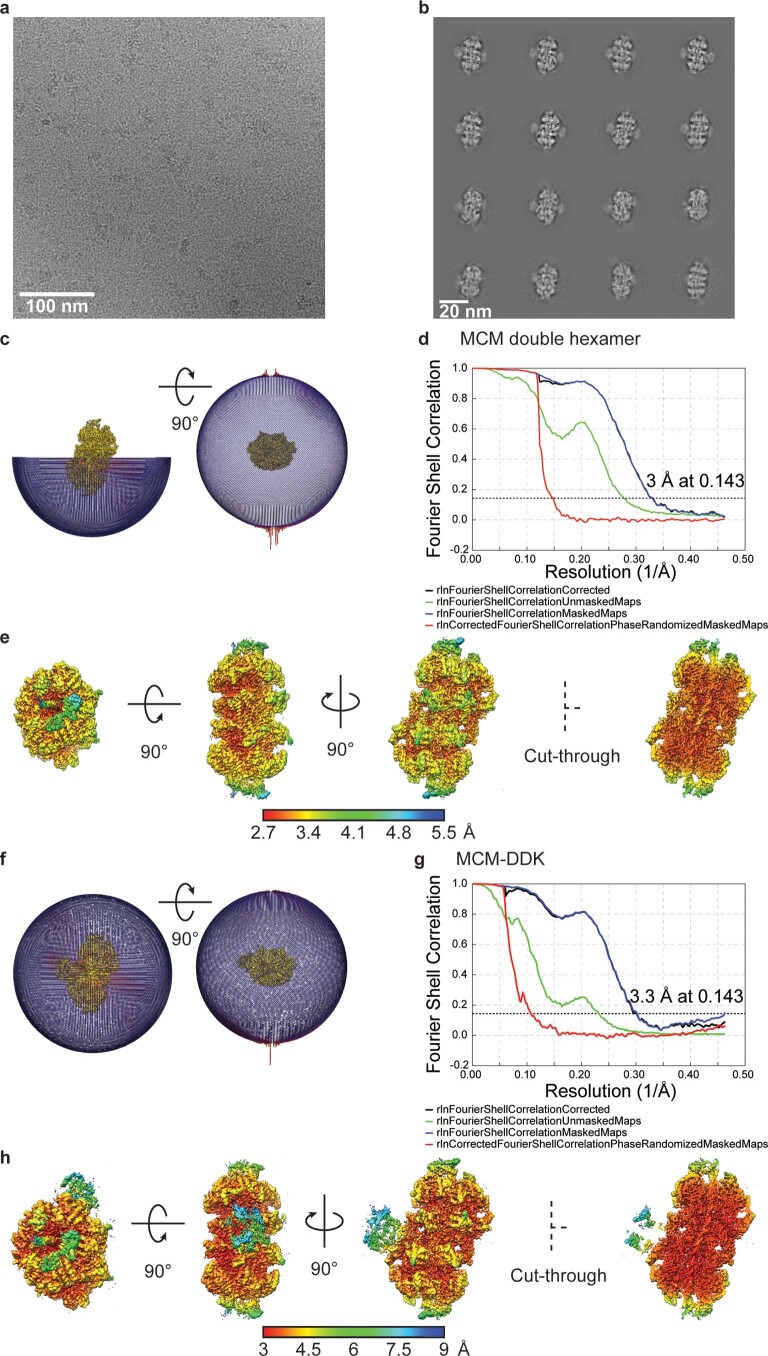
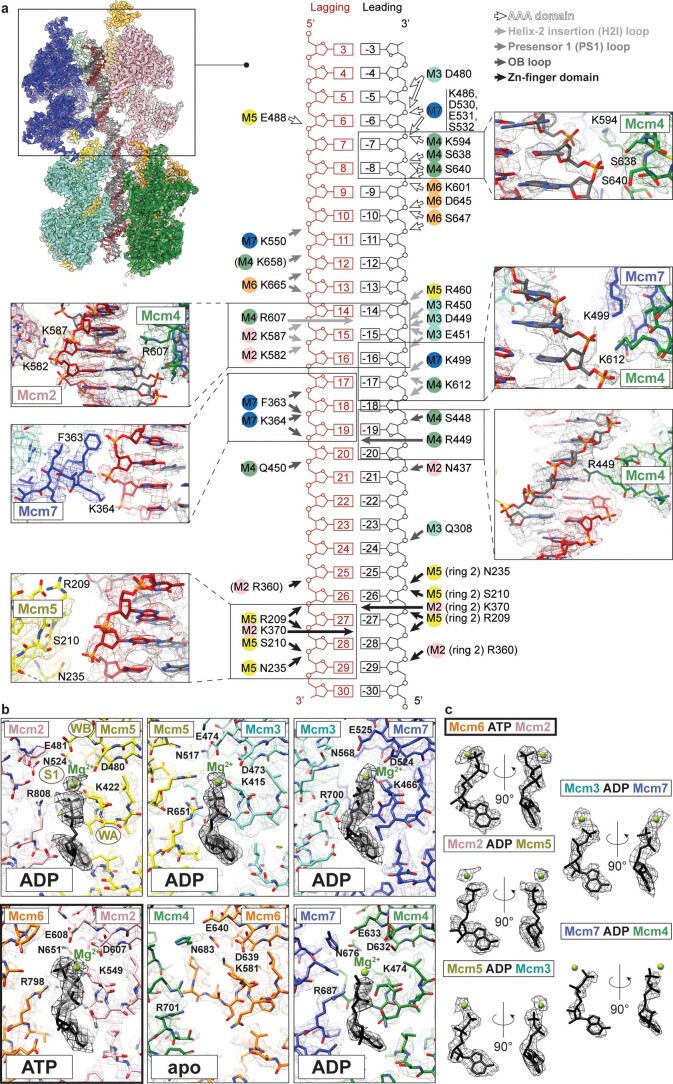
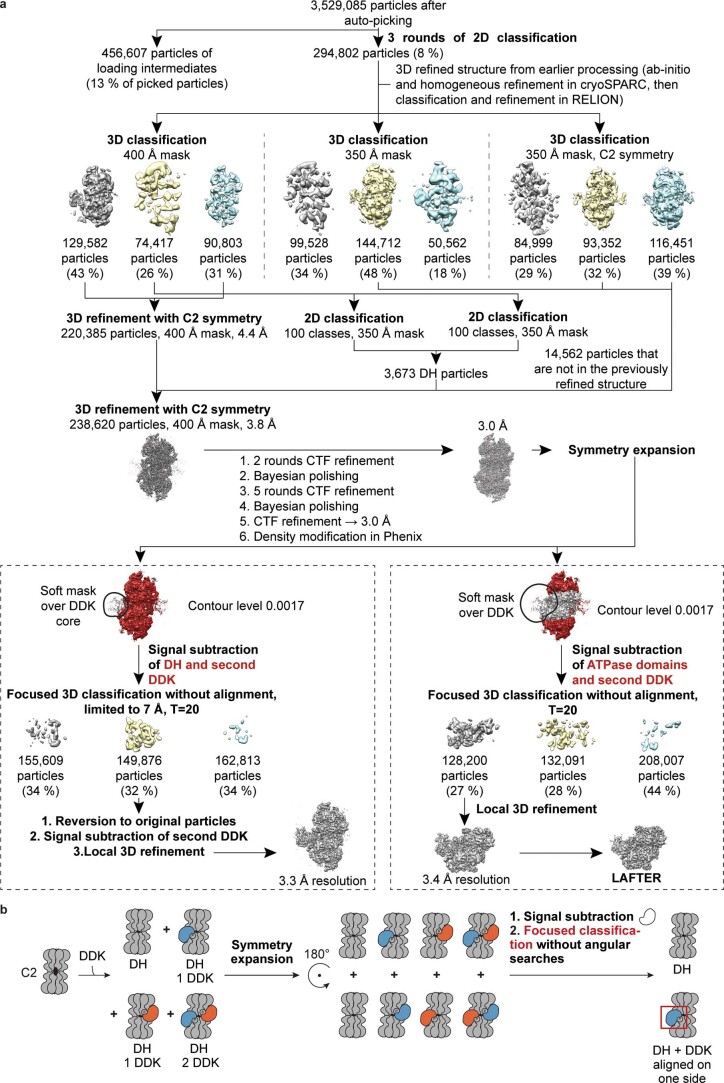
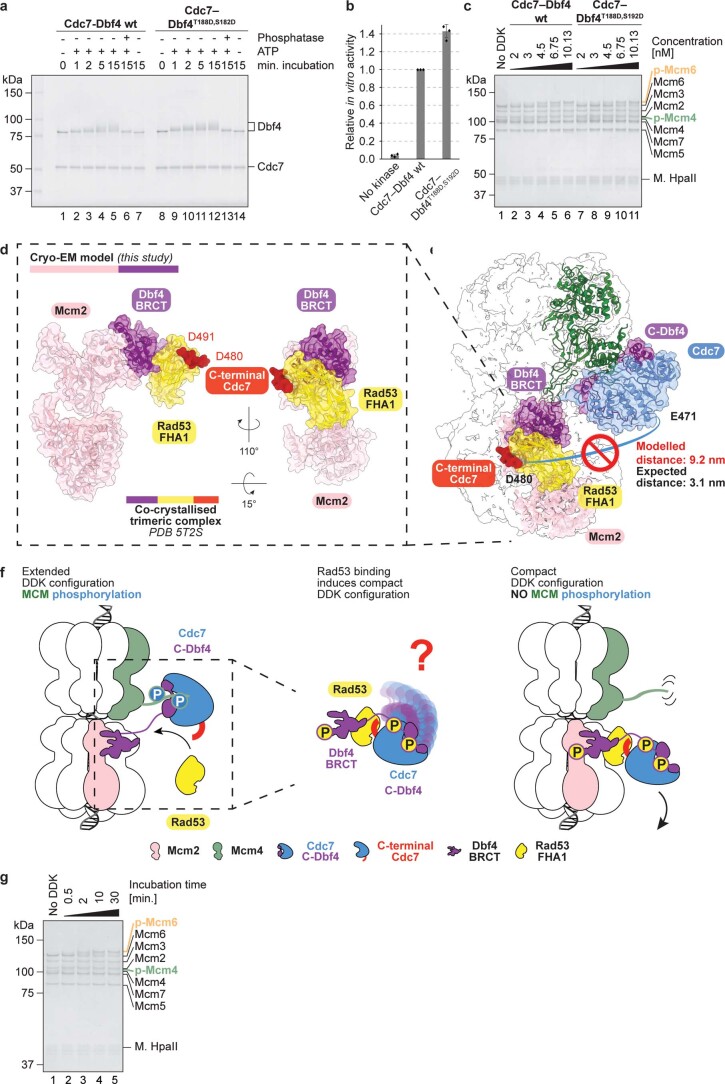
Loading of the eukaryotic replicative helicase onto replication origins involves two MCM hexamers forming a double hexamer (DH) around duplex DNA. During S phase, helicase activation requires MCM phosphorylation by Dbf4-dependent kinase (DDK), comprising Cdc7 and Dbf4. DDK selectively phosphorylates loaded DHs, but how such fidelity is achieved is unknown. Here, we determine the cryogenic electron microscopy structure of Saccharomyces cerevisiae DDK in the act of phosphorylating a DH. DDK docks onto one MCM ring and phosphorylates the opposed ring. Truncation of the Dbf4 docking domain abrogates DH phosphorylation, yet Cdc7 kinase activity is unaffected. Late origin firing is blocked in response to DNA damage via Dbf4 phosphorylation by the Rad53 checkpoint kinase. DDK phosphorylation by Rad53 impairs DH phosphorylation by blockage of DDK binding to DHs, and also interferes with the Cdc7 active site. Our results explain the structural basis and regulation of the selective phosphorylation of DNA-loaded MCM DHs, which supports bidirectional replication.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
