

Insulin Resistance: From Mechanisms to Therapeutic Strategies
- PMID: 34965646
- PMCID: PMC8831809
- DOI: 10.4093/dmj.2021.0280
Insulin Resistance: From Mechanisms to Therapeutic Strategies
Abstract
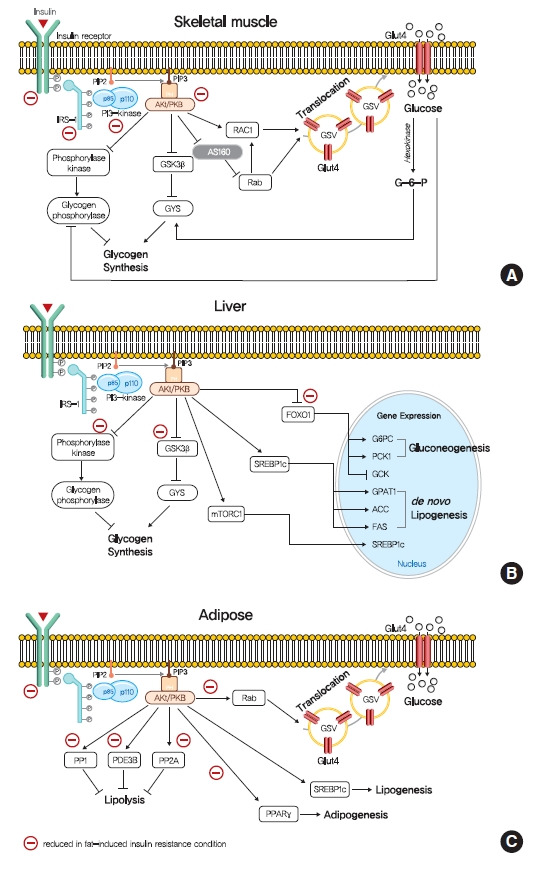
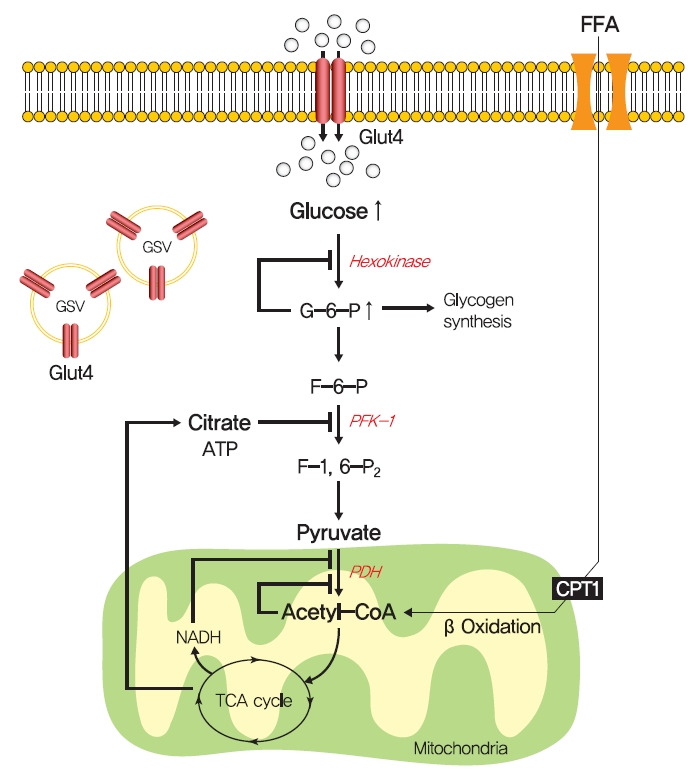
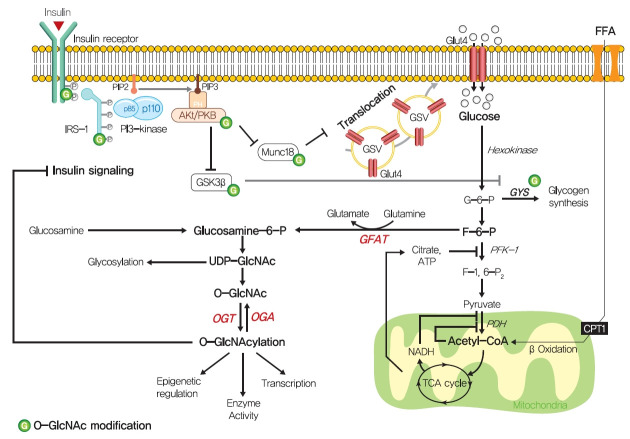
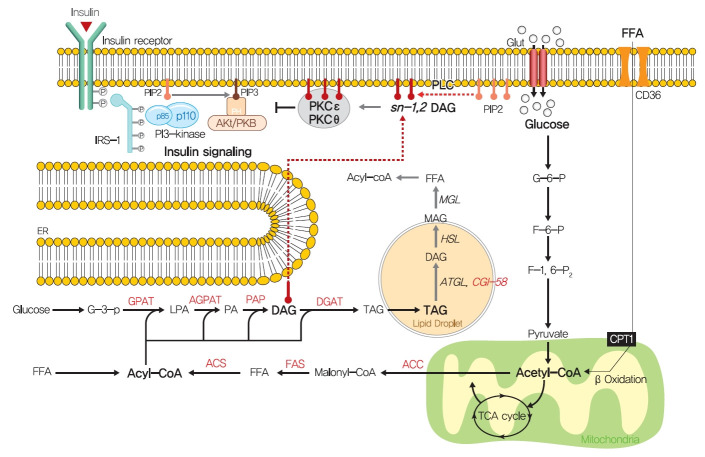
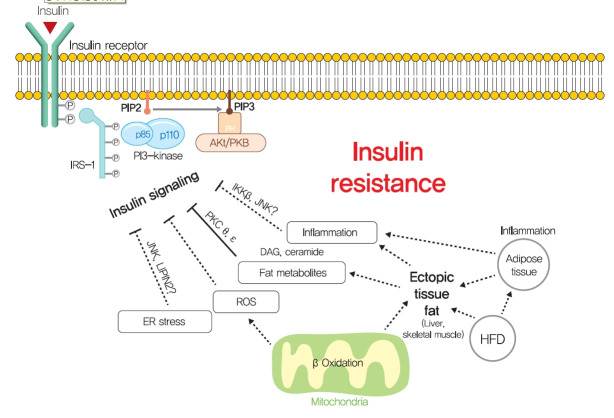
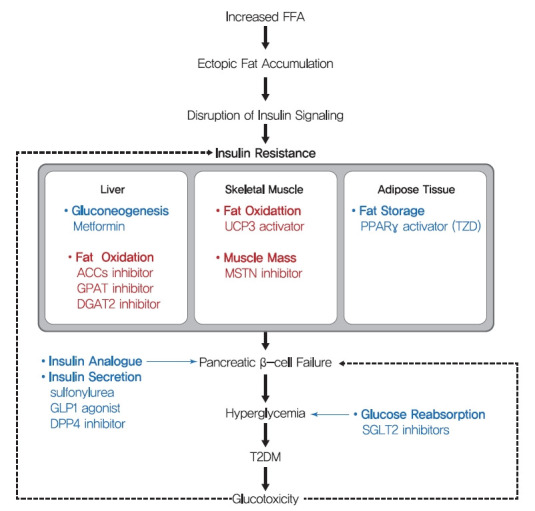
Insulin resistance is the pivotal pathogenic component of many metabolic diseases, including type 2 diabetes mellitus, and is defined as a state of reduced responsiveness of insulin-targeting tissues to physiological levels of insulin. Although the underlying mechanism of insulin resistance is not fully understood, several credible theories have been proposed. In this review, we summarize the functions of insulin in glucose metabolism in typical metabolic tissues and describe the mechanisms proposed to underlie insulin resistance, that is, ectopic lipid accumulation in liver and skeletal muscle, endoplasmic reticulum stress, and inflammation. In addition, we suggest potential therapeutic strategies for addressing insulin resistance.
Keywords: Diabetes mellitus, type 2; Insulin resistance; Metabolic syndrome; Therapeutics.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
