

A catalogue of 1,167 genomes from the human gut archaeome
- PMID: 34969981
- PMCID: PMC8727293
- DOI: 10.1038/s41564-021-01020-9
A catalogue of 1,167 genomes from the human gut archaeome
Erratum in
-
Publisher Correction: A catalogue of 1,167 genomes from the human gut archaeome.Nat Microbiol. 2022 Feb;7(2):339. doi: 10.1038/s41564-022-01061-8. Nat Microbiol. 2022. PMID: 35013594 Free PMC article. No abstract available.
Abstract
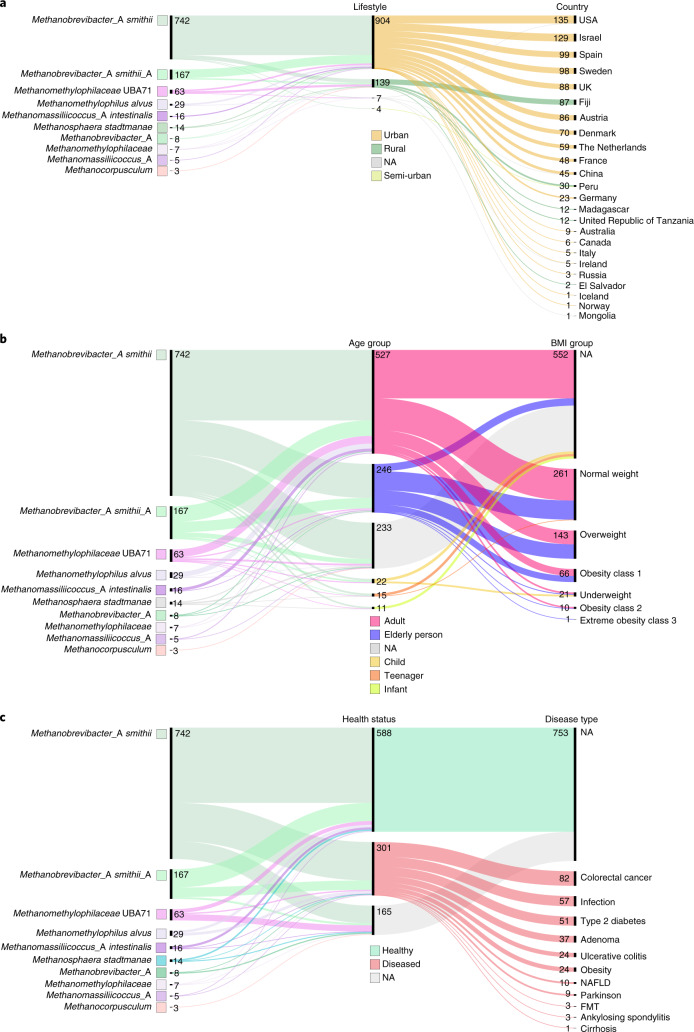
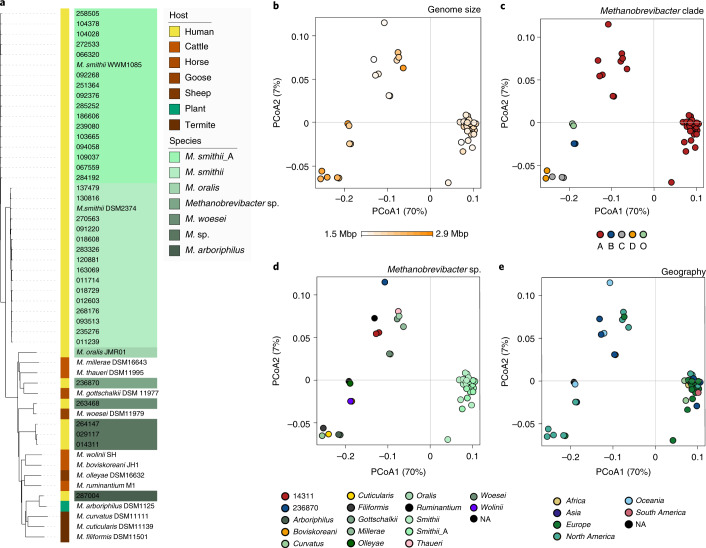
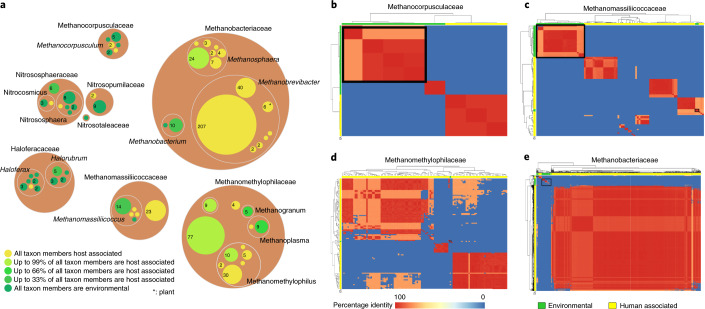
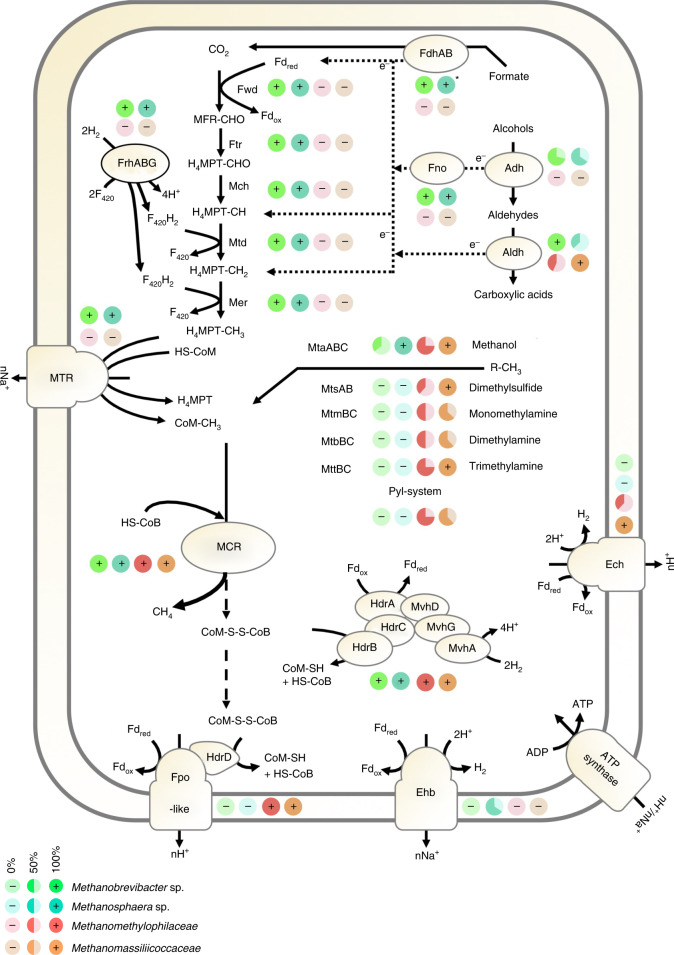
The human gut microbiome plays an important role in health, but its archaeal diversity remains largely unexplored. In the present study, we report the analysis of 1,167 nonredundant archaeal genomes (608 high-quality genomes) recovered from human gastrointestinal tract, sampled across 24 countries and rural and urban populations. We identified previously undescribed taxa including 3 genera, 15 species and 52 strains. Based on distinct genomic features, we justify the split of the Methanobrevibacter smithii clade into two separate species, with one represented by the previously undescribed 'Candidatus Methanobrevibacter intestini'. Patterns derived from 28,581 protein clusters showed significant associations with sociodemographic characteristics such as age groups and lifestyle. We additionally show that archaea are characterized by specific genomic and functional adaptations to the host and carry a complex virome. Our work expands our current understanding of the human archaeome and provides a large genome catalogue for future analyses to decipher its impact on human physiology.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












Comment in
-
The human archaeome in focus.Nat Microbiol. 2022 Jan;7(1):10-11. doi: 10.1038/s41564-021-01031-6. Nat Microbiol. 2022. PMID: 34969980 No abstract available.
References
-
- Moissl-Eichinger, C. et al. Archaea are interactive components of complex microbiomes. Trends Microbiol. 26, 70–85 (2018). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
