

Immune Complex Formation Is Associated With Loss of Tolerance and an Antibody Response to Both Drug and Target
- PMID: 34970265
- PMCID: PMC8712722
- DOI: 10.3389/fimmu.2021.782788
Immune Complex Formation Is Associated With Loss of Tolerance and an Antibody Response to Both Drug and Target
Abstract
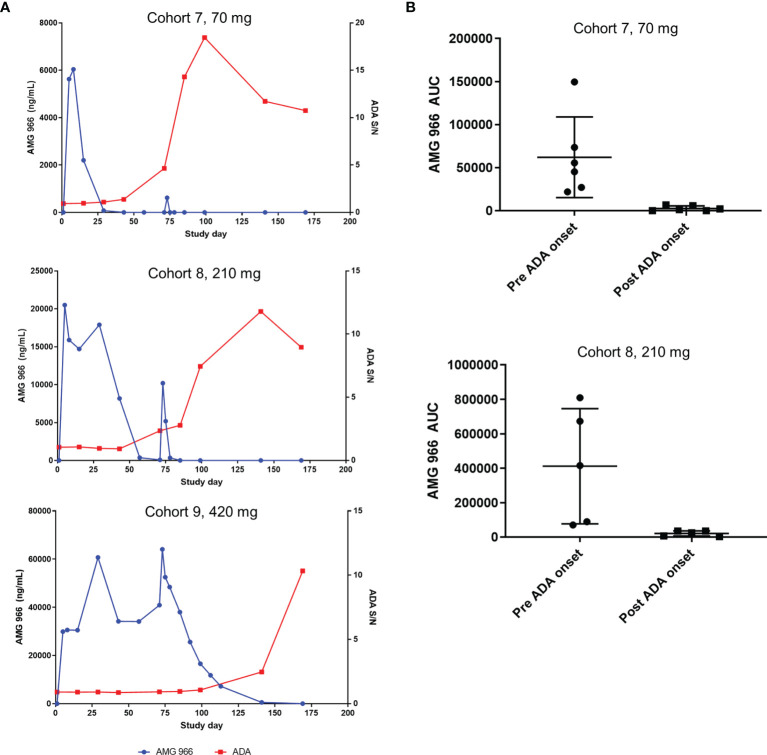
AMG 966 is a bi-specific, heteroimmunoglobulin molecule that binds both tumor necrosis factor alpha (TNFα) and TNF-like ligand 1A (TL1A). In a first-in-human clinical study in healthy volunteers, AMG 966 elicited anti-drug antibodies (ADA) in 53 of 54 subjects (98.1%), despite a paucity of T cell epitopes observed in T cell assays. ADA were neutralizing and bound to all domains of AMG 966. Development of ADA correlated with loss of exposure. In vitro studies demonstrated that at certain drug-to-target ratios, AMG 966 forms large immune complexes with TNFα and TL1A, partially restoring the ability of the aglycosylated Fc domain to bind FcγRIa and FcγRIIa, leading to the formation of ADA. In addition to ADA against AMG 966, antibodies to endogenous TNFα were also detected in the sera of subjects dosed with AMG 966. This suggests that the formation of immune complexes between a therapeutic and target can cause loss of tolerance and elicit an antibody response against the target.
Keywords: AMG 966; TL1A; TNFα; anti-drug antibodies; immune complexes; immunogenicity; inflammatory bowel disease; tolerance.
Copyright © 2021 Kroenke, Barger, Hu, Miller, Kalenian, He, Hsu, Bartley, Chow, Teixeira dos Santos, Sullivan, Cheng, Parnes, Padaki, Kuhns and Mytych.
Conflict of interest statement
All authors were employees of Amgen during the time the study was being conducted. The authors declare this study received funding from Amgen. The funder was responsible for study design, interpretation of data, and the writing of this article. AMG 966 is disclosed in patent application WO2017/106383.
Figures







Similar articles
-
Immunogenicity of TNF-Inhibitors.Front Immunol. 2020 Feb 26;11:312. doi: 10.3389/fimmu.2020.00312. eCollection 2020. Front Immunol. 2020. PMID: 32174918 Free PMC article. Review.
-
Characterization and root cause analysis of immunogenicity to pasotuxizumab (AMG 212), a prostate-specific membrane antigen-targeting bispecific T-cell engager therapy.Front Immunol. 2023 Oct 23;14:1261070. doi: 10.3389/fimmu.2023.1261070. eCollection 2023. Front Immunol. 2023. PMID: 37942314 Free PMC article.
-
Addressing soluble target interference in the development of a functional assay for the detection of neutralizing antibodies against a BCMA-CD3 bispecific antibody.J Immunol Methods. 2019 Nov;474:112642. doi: 10.1016/j.jim.2019.112642. Epub 2019 Aug 7. J Immunol Methods. 2019. PMID: 31400410
-
Characterization of anti-drug antibody responses to the T-cell engaging bispecific antibody cibisatamab to understand the impact on exposure.Front Immunol. 2024 May 31;15:1406353. doi: 10.3389/fimmu.2024.1406353. eCollection 2024. Front Immunol. 2024. PMID: 38881900 Free PMC article. Clinical Trial.
-
The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment With Monoclonal Antibodies.Front Immunol. 2020 Aug 18;11:1951. doi: 10.3389/fimmu.2020.01951. eCollection 2020. Front Immunol. 2020. PMID: 33013848 Free PMC article. Review.
Cited by
-
HLAII peptide presentation of infliximab increases when complexed with TNF.Front Immunol. 2022 Sep 13;13:932252. doi: 10.3389/fimmu.2022.932252. eCollection 2022. Front Immunol. 2022. PMID: 36177046 Free PMC article.
-
Development of High-Titer Antidrug Antibodies in a Phase 1b/2a Infant Clesrovimab Trial Are Associated With RSV Exposure Beyond Day 150.J Infect Dis. 2025 Mar 17;231(3):e488-e496. doi: 10.1093/infdis/jiae582. J Infect Dis. 2025. PMID: 39590882 Free PMC article. Clinical Trial.
-
Therapeutic drug monitoring and immunogenetic factors associated with the use of adalimumab in Crohn's disease patients.Int J Immunopathol Pharmacol. 2025 Jan-Dec;39:3946320251319379. doi: 10.1177/03946320251319379. Int J Immunopathol Pharmacol. 2025. PMID: 39959979 Free PMC article.
-
Targeting TNF/TNFR superfamilies in immune-mediated inflammatory diseases.J Exp Med. 2024 Nov 4;221(11):e20240806. doi: 10.1084/jem.20240806. Epub 2024 Sep 19. J Exp Med. 2024. PMID: 39297883 Free PMC article. Review.
-
Immunogenicity risk assessment and mitigation for engineered antibody and protein therapeutics.Nat Rev Drug Discov. 2024 Dec;23(12):898-913. doi: 10.1038/s41573-024-01051-x. Epub 2024 Oct 18. Nat Rev Drug Discov. 2024. PMID: 39424922 Review.
References
-
- D'Haens G, Van Deventer S, Van Hogezand R, Chalmers D, Kothe C, Baert F, et al. . Endoscopic and Histological Healing With Infliximab Anti-Tumor Necrosis Factor Antibodies in Crohn's Disease: A European Multicenter Trial. Gastroenterology (1999) 116(5):1029–34. doi: 10.1016/s0016-5085(99)70005-3 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials