

FcγRIIB potentiates differentiation of myeloid-derived suppressor cells to mediate tumor immunoescape
- PMID: 34976216
- PMCID: PMC8692894
- DOI: 10.7150/thno.66575
FcγRIIB potentiates differentiation of myeloid-derived suppressor cells to mediate tumor immunoescape
Abstract
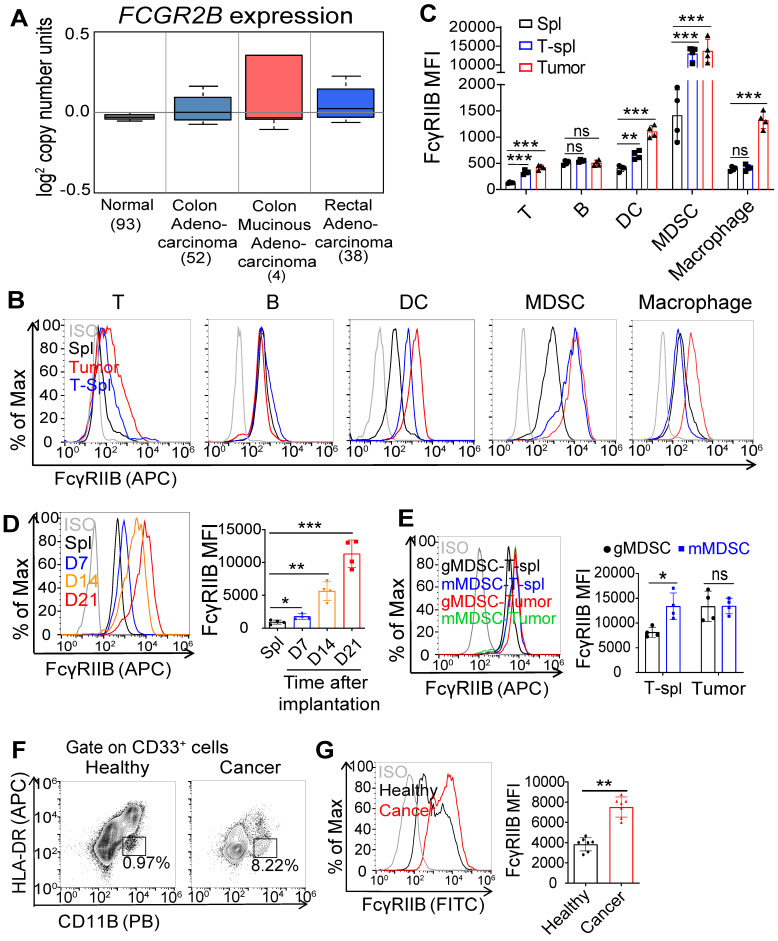
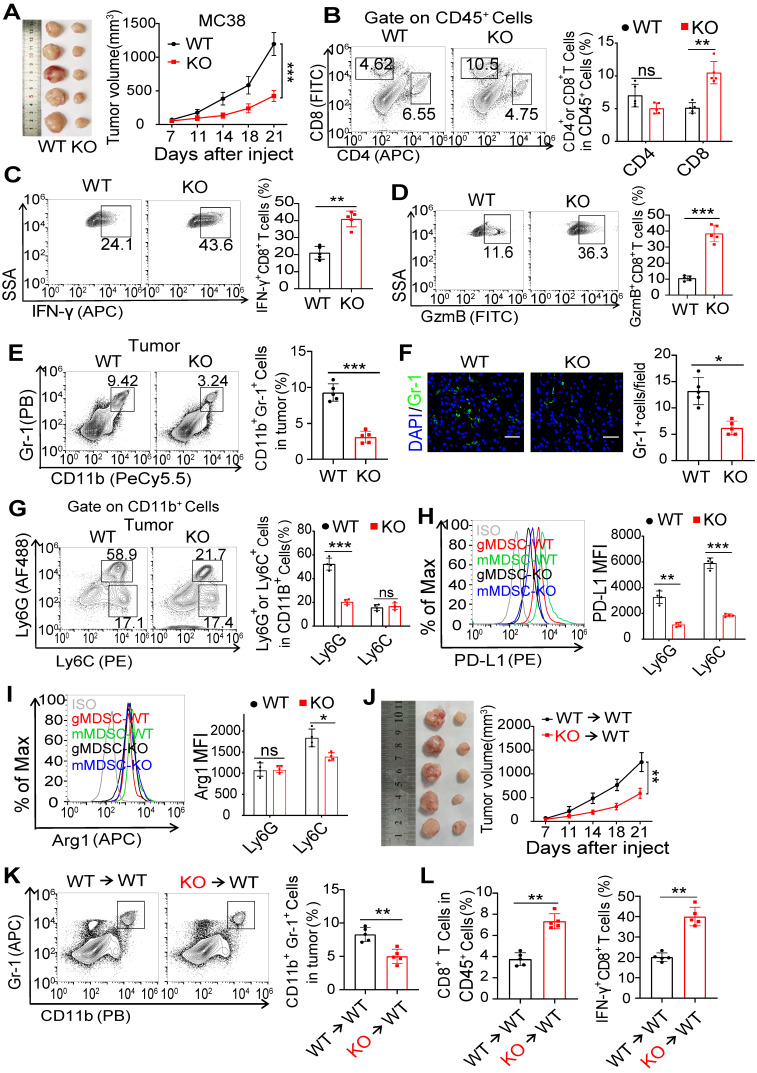
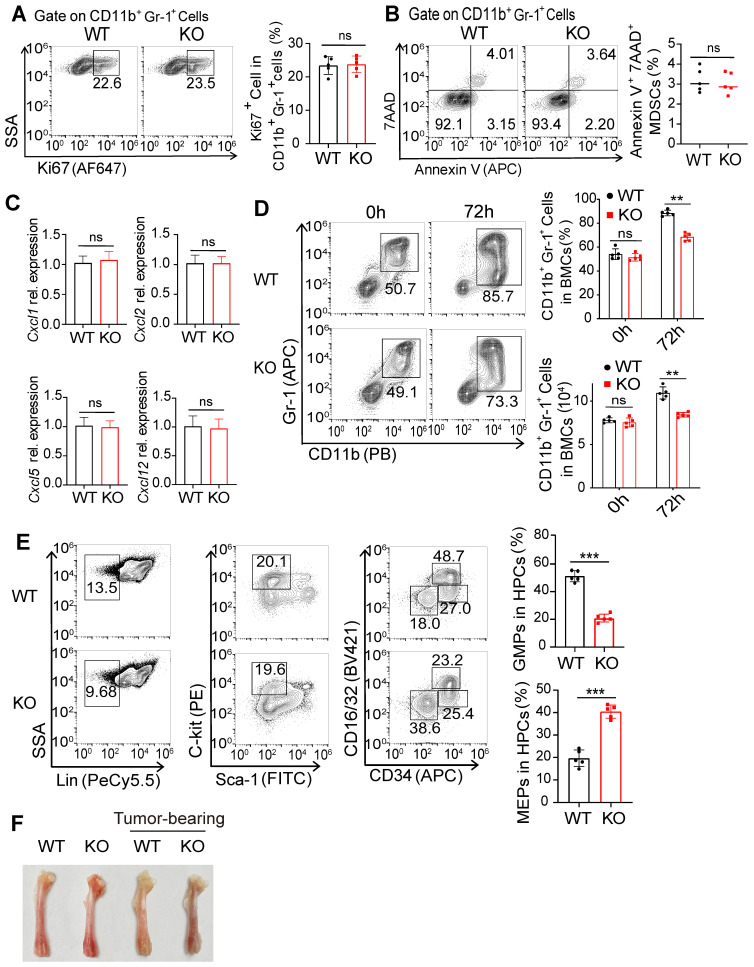
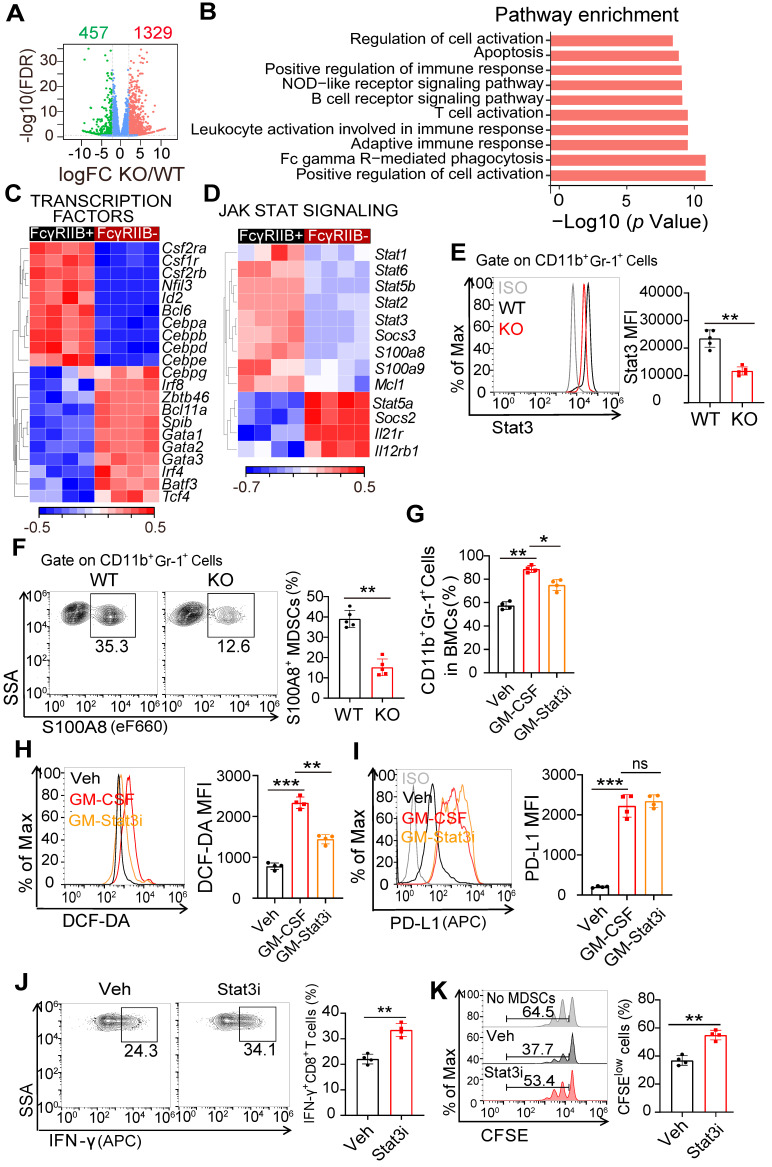
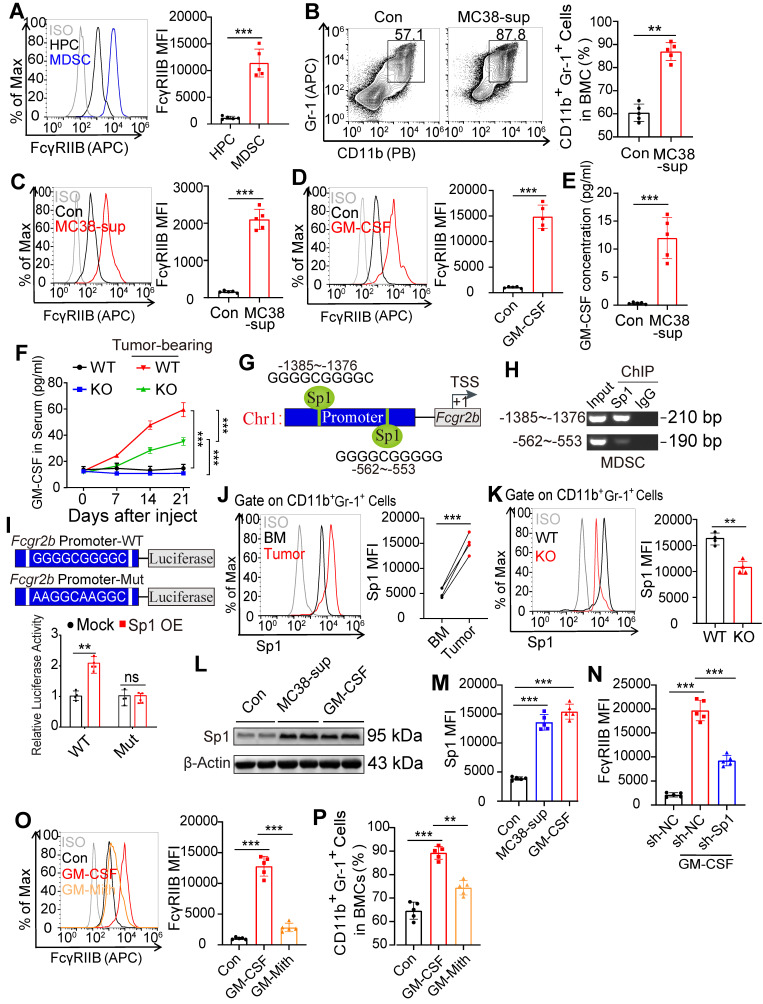
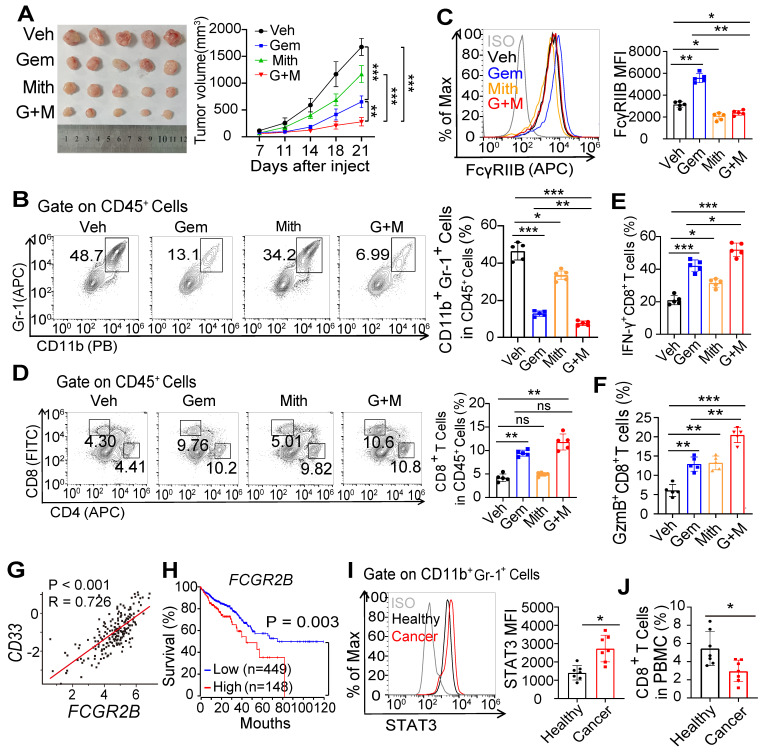
Background: FcγRIIB, the sole inhibitory receptor of the Fc gamma receptor family, plays pivotal roles in innate and adaptive immune responses. However, the expression and function of FcγRIIB in myeloid-derived suppressor cells (MDSCs) remains unknown. This study aimed to investigate whether and how FcγRIIB regulates the immunosuppressive activity of MDSCs during cancer development. Methods: The MC38 and B16-F10 tumor-bearing mouse models were established to investigate the role of FcγRIIB during tumor progression. FcγRIIB-deficient mice, adoptive cell transfer, mRNA-sequencing and flow cytometry analysis were used to assess the role of FcγRIIB on immunosuppressive activity and differentiation of MDSCs. Results: Here we show that FcγRIIB was upregulated in tumor-infiltrated MDSCs. FcγRIIB-deficient mice showed decreased accumulation of MDSCs in the tumor microenvironment (TME) compared with wild-type mice. FcγRIIB was required for the differentiation and immunosuppressive activity of MDSCs. Mechanistically, tumor cell-derived granulocyte-macrophage colony stimulating factor (GM-CSF) increased the expression of FcγRIIB on hematopoietic progenitor cells (HPCs) by activating specificity protein 1 (Sp1), subsequently FcγRIIB promoted the generation of MDSCs from HPCs via Stat3 signaling. Furthermore, blockade of Sp1 dampened MDSC differentiation and infiltration in the TME and enhanced the anti-tumor therapeutic efficacy of gemcitabine. Conclusion: These results uncover an unrecognized regulatory role of the FcγRIIB in abnormal differentiation of MDSCs during cancer development and suggest a potential therapeutic target for anti-tumor therapy.
Keywords: Fc gamma receptor IIB; Sp1 signaling; anti-tumor therapy; granulocyte-macrophage colony stimulating factor; immunosuppression; myeloid-derived suppressor cells; tumor microenvironment.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures

References
-
- Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH. et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14(-)/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136:35–45. - PMC - PubMed
-
- Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
