

Time-controlled and muscle-specific CRISPR/Cas9-mediated deletion of CTG-repeat expansion in the DMPK gene
- PMID: 34976437
- PMCID: PMC8693309
- DOI: 10.1016/j.omtn.2021.11.024
Time-controlled and muscle-specific CRISPR/Cas9-mediated deletion of CTG-repeat expansion in the DMPK gene
Abstract
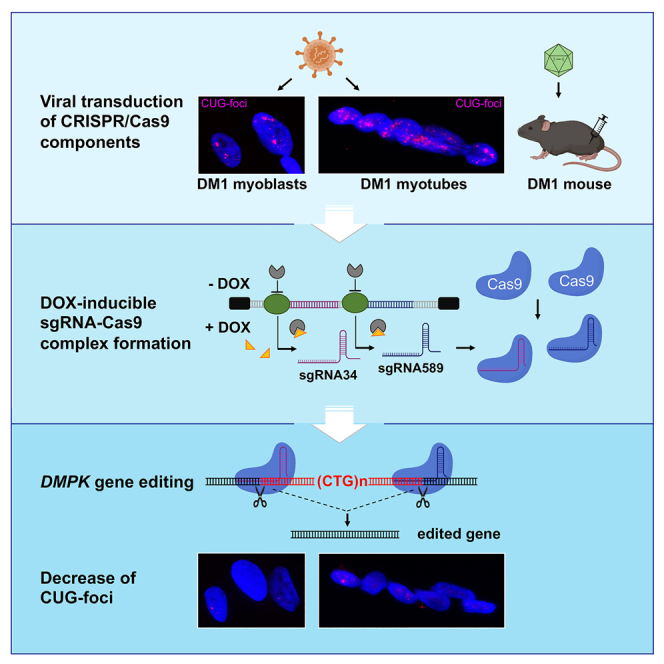
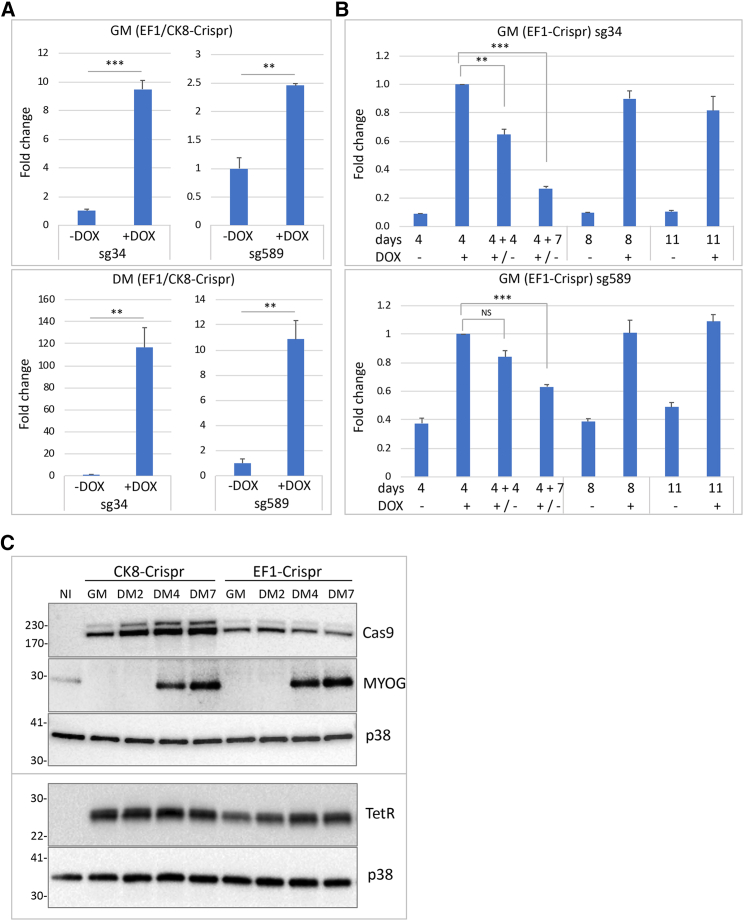
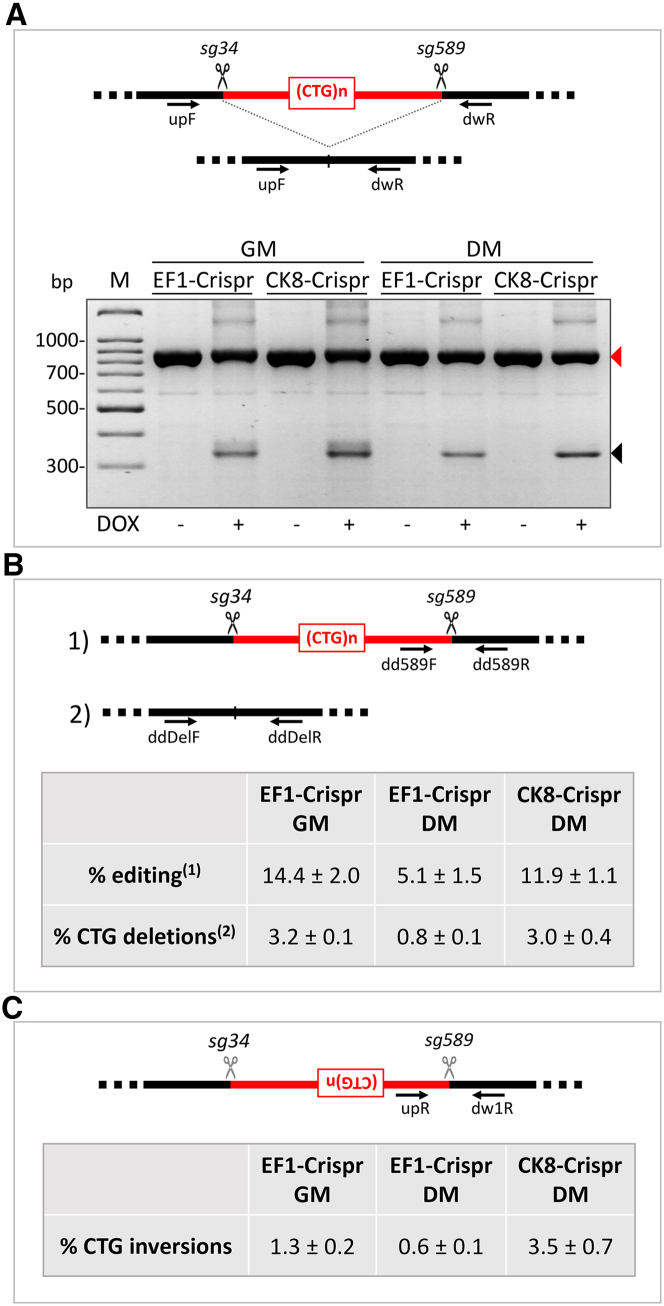
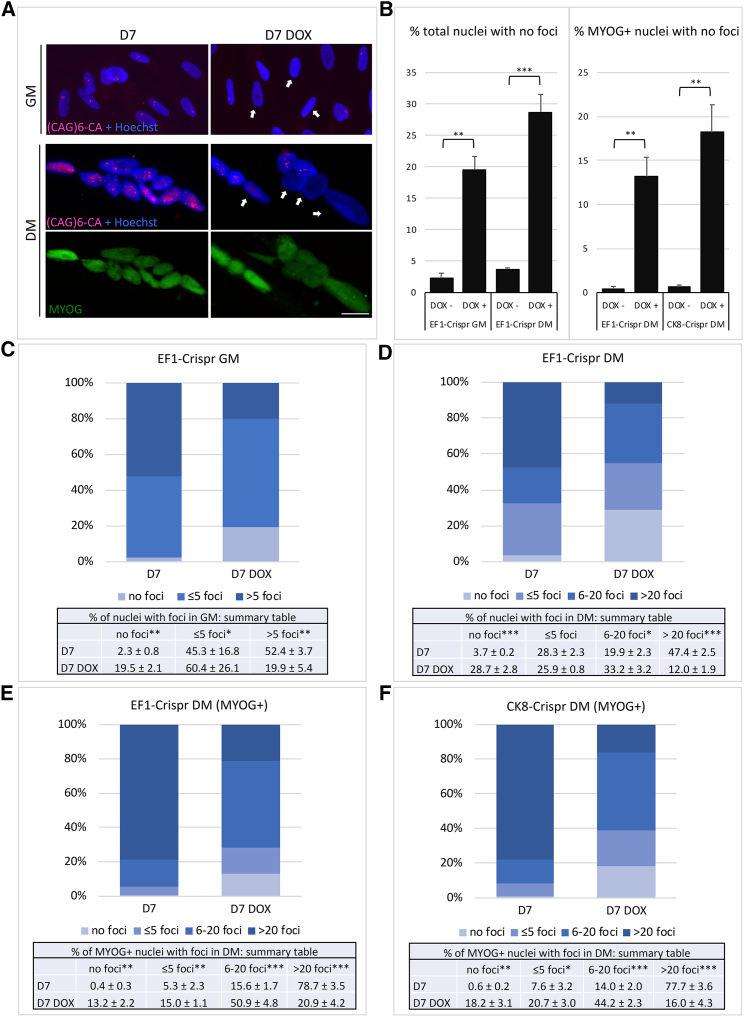
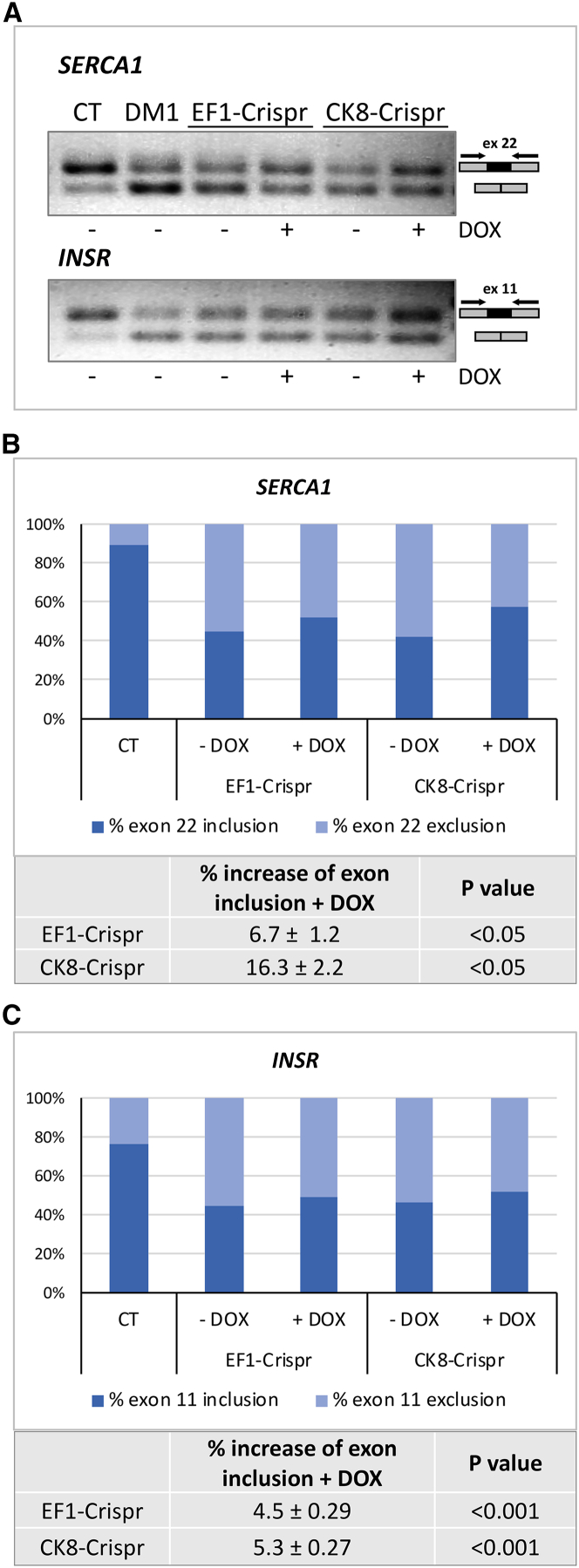
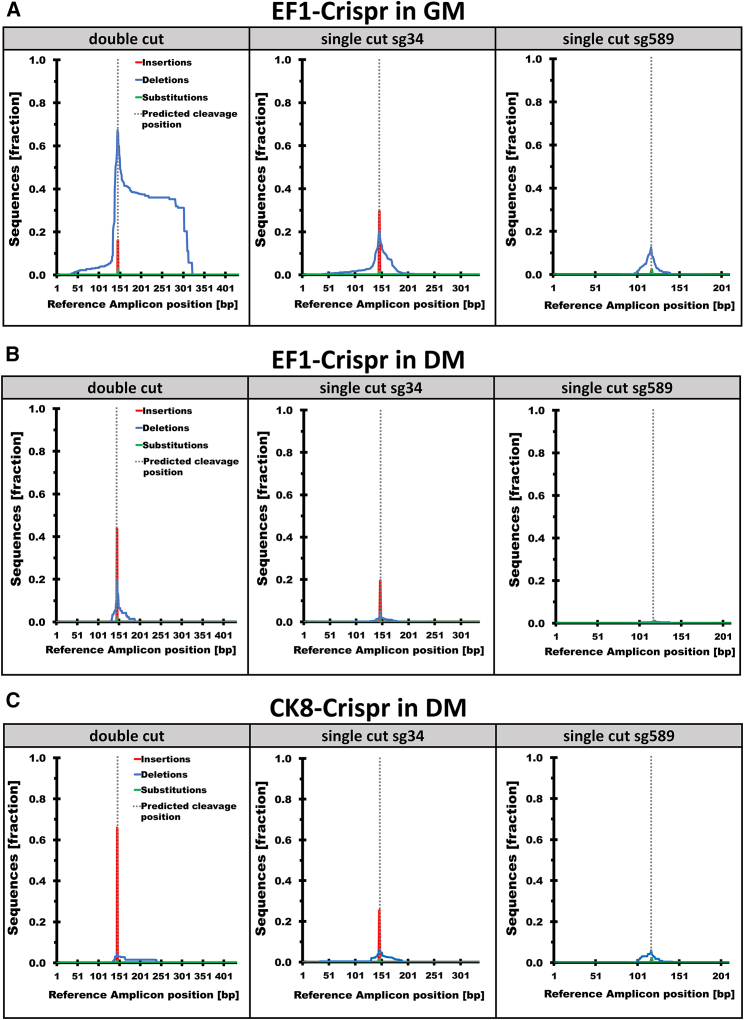
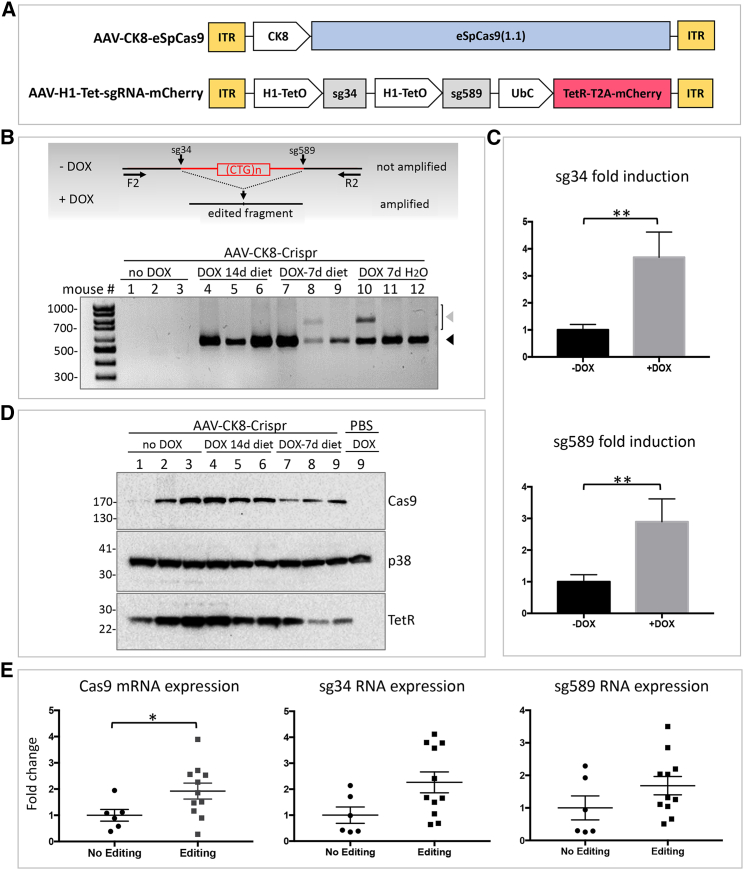
CRISPR/Cas9-mediated therapeutic gene editing is a promising technology for durable treatment of incurable monogenic diseases such as myotonic dystrophies. Gene-editing approaches have been recently applied to in vitro and in vivo models of myotonic dystrophy type 1 (DM1) to delete the pathogenic CTG-repeat expansion located in the 3' untranslated region of the DMPK gene. In DM1-patient-derived cells removal of the expanded repeats induced beneficial effects on major hallmarks of the disease with reduction in DMPK transcript-containing ribonuclear foci and reversal of aberrant splicing patterns. Here, we set out to excise the triplet expansion in a time-restricted and cell-specific fashion to minimize the potential occurrence of unintended events in off-target genomic loci and select for the target cell type. To this aim, we employed either a ubiquitous promoter-driven or a muscle-specific promoter-driven Cas9 nuclease and tetracycline repressor-based guide RNAs. A dual-vector approach was used to deliver the CRISPR/Cas9 components into DM1 patient-derived cells and in skeletal muscle of a DM1 mouse model. In this way, we obtained efficient and inducible gene editing both in proliferating cells and differentiated post-mitotic myocytes in vitro as well as in skeletal muscle tissue in vivo.
Keywords: CRISPR/Cas9; CTG repeats; DM1; DMSXL mouse model; gene editing; gene therapy; myotonic dystrophy; skeletal muscle.
© 2021 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
