

Glutaric Acidemia, Pathogenesis and Nutritional Therapy
- PMID: 34977106
- PMCID: PMC8714794
- DOI: 10.3389/fnut.2021.704984
Glutaric Acidemia, Pathogenesis and Nutritional Therapy
Abstract
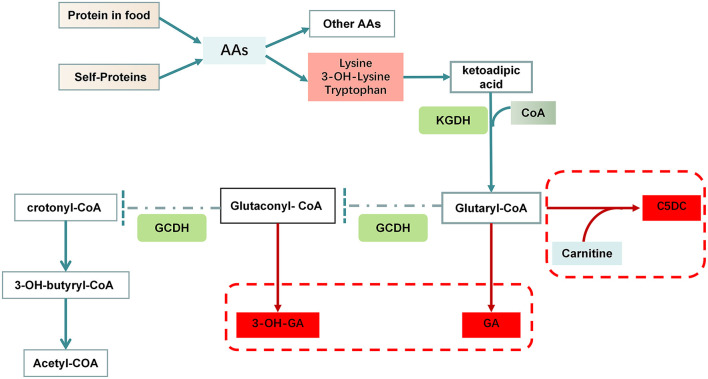
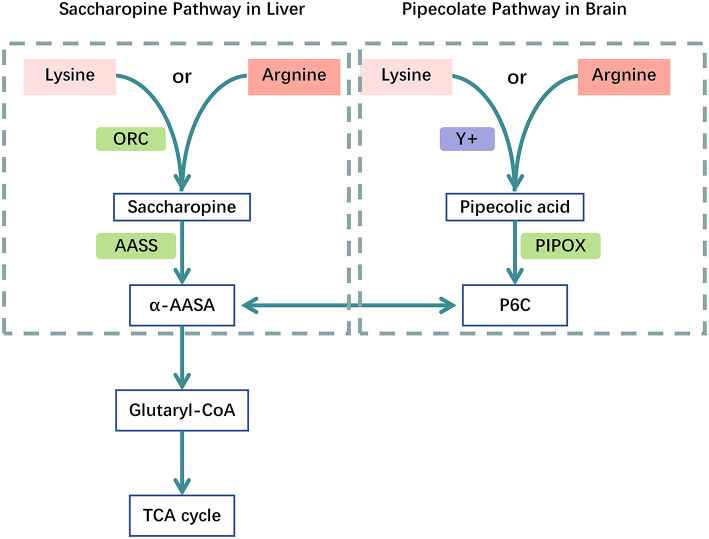
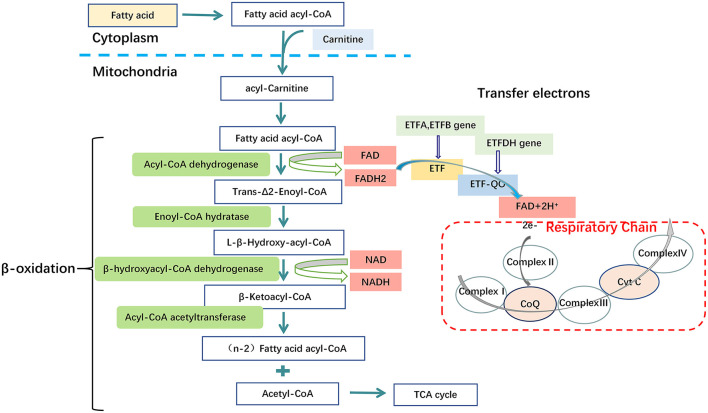
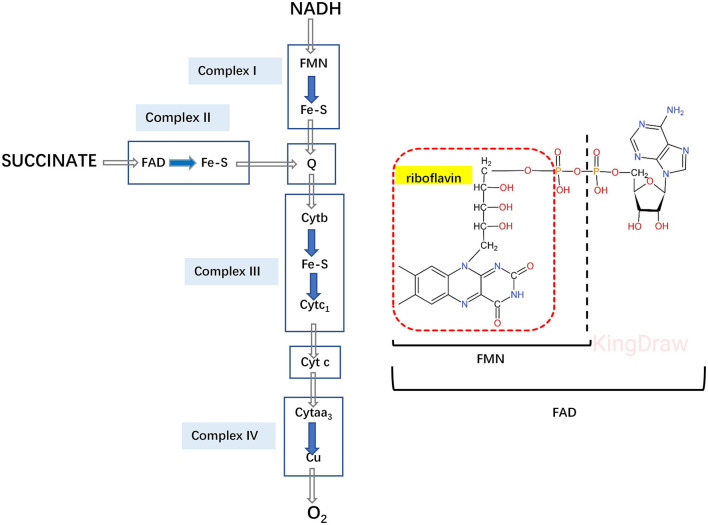
Glutaric acidemia (GA) are heterogeneous, genetic diseases that present with specific catabolic deficiencies of amino acid or fatty acid metabolism. The disorders can be divided into type I and type II by the occurrence of different types of recessive mutations of autosomal, metabolically important genes. Patients of glutaric acidemia type I (GA-I) if not diagnosed very early in infanthood, experience irreversible neurological injury during an encephalopathic crisis in childhood. If diagnosed early the disorder can be treated successfully with a combined metabolic treatment course that includes early catabolic emergency treatment and long-term maintenance nutrition therapy. Glutaric acidemia type II (GA- II) patients can present clinically with hepatomegaly, non-ketotic hypoglycemia, metabolic acidosis, hypotonia, and in neonatal onset cardiomyopathy. Furthermore, it features adult-onset muscle-related symptoms, including weakness, fatigue, and myalgia. An early diagnosis is crucial, as both types can be managed by simple nutraceutical supplementation. This review discusses the pathogenesis of GA and its nutritional management practices, and aims to promote understanding and management of GA. We will provide a detailed summary of current clinical management strategies of the glutaric academia disorders and highlight issues of nutrition therapy principles in emergency settings and outline some specific cases.
Keywords: Glutaric acidemia; genetic disorders; maintenance therapy; nutrition therapy; pathogenesis.
Copyright © 2021 Li, Yang, Feng, Zhao, Su, Liu, Men, Huang, Körner and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Glutaric Acidemia Type 1: Diagnosis, Clinical features, and Outcome in a Portuguese Cohort.Endocr Metab Immune Disord Drug Targets. 2023 Sep 14. doi: 10.2174/1871530323666230914122946. Online ahead of print. Endocr Metab Immune Disord Drug Targets. 2023. PMID: 37711119
-
A novel electron transfer flavoprotein dehydrogenase (ETFDH) gene mutation identified in a newborn with glutaric acidemia type II: a case report of a Chinese family.BMC Med Genet. 2020 May 11;21(1):98. doi: 10.1186/s12881-020-00995-2. BMC Med Genet. 2020. PMID: 32393189 Free PMC article.
-
Early Diagnosed and Treated Glutaric Acidemia Type 1 Female Presenting with Subependymal Nodules in Adulthood.JIMD Rep. 2018;40:85-90. doi: 10.1007/8904_2017_66. Epub 2017 Nov 1. JIMD Rep. 2018. PMID: 29086383 Free PMC article.
-
Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision.J Inherit Metab Dis. 2017 Jan;40(1):75-101. doi: 10.1007/s10545-016-9999-9. Epub 2016 Nov 16. J Inherit Metab Dis. 2017. PMID: 27853989 Review.
-
A role of astrocytes in mediating postnatal neurodegeneration in Glutaric acidemia-type 1.FEBS Lett. 2015 Nov 14;589(22):3492-7. doi: 10.1016/j.febslet.2015.09.010. Epub 2015 Sep 26. FEBS Lett. 2015. PMID: 26409499 Review.
Cited by
-
Glutaric Aciduria Presenting With an Acute Encephalitic Crisis: A Case Report.Cureus. 2024 Jul 30;16(7):e65722. doi: 10.7759/cureus.65722. eCollection 2024 Jul. Cureus. 2024. PMID: 39211641 Free PMC article.
-
Spectrum of congenital anomalies of the kidney and urinary tract (CAKUT) including renal parenchymal malformations during fetal life and the implementation of prenatal exome sequencing (WES).Arch Gynecol Obstet. 2024 Jun;309(6):2613-2622. doi: 10.1007/s00404-023-07165-8. Epub 2023 Aug 3. Arch Gynecol Obstet. 2024. PMID: 37535131 Free PMC article.
-
Incidence and prevalence of 121 rare diseases in China: Current status and challenges: 2022 revision.Intractable Rare Dis Res. 2022 Aug;11(3):96-104. doi: 10.5582/irdr.2022.01093. Intractable Rare Dis Res. 2022. PMID: 36200031 Free PMC article. Review.
-
Neonatal necrotizing enterocolitis complicated by glutaric acidemia type II: a case report.Front Pediatr. 2025 Feb 3;13:1392927. doi: 10.3389/fped.2025.1392927. eCollection 2025. Front Pediatr. 2025. PMID: 39963347 Free PMC article.
-
Two novel compound heterozygous variants of the GCDH gene in two Chinese families with glutaric acidaemia type I identified by high-throughput sequencing and a literature review.Mol Genet Genomics. 2023 May;298(3):603-614. doi: 10.1007/s00438-023-02002-8. Epub 2023 Mar 11. Mol Genet Genomics. 2023. PMID: 36906724 Review.
References
Publication types
LinkOut - more resources
Full Text Sources