

CUBN gene mutations may cause focal segmental glomerulosclerosis (FSGS) in children
- PMID: 34979989
- PMCID: PMC8725476
- DOI: 10.1186/s12882-021-02654-x
CUBN gene mutations may cause focal segmental glomerulosclerosis (FSGS) in children
Abstract
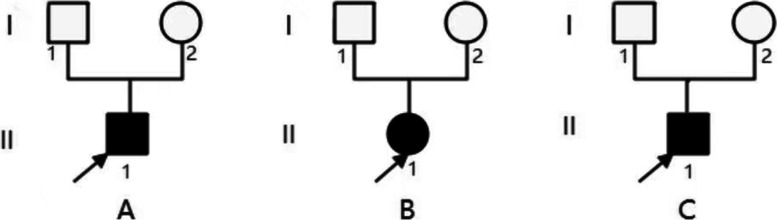
Background: Imerslund-Gräsbeck Syndrome (IGS) is mainly caused by CUBN gene biallelic mutations. Proteinuria accompanies IGS specific symptoms in about half of the patients, isolated proteinuria is rarely reported. Here we present 3 patients with isolated proteinuria and focal segmental glomerulosclerosis (FSGS) caused by CUBN gene biallelic pathogenic variants.
Method: Whole exome sequencing was performed on three children with isolated proteinuria. CUBN gene biallelic pathogenic variants were found and then verified by sanger sequencing. Their clinical, pathological and molecular genetic characteristics were analyzed and correlated accordingly.
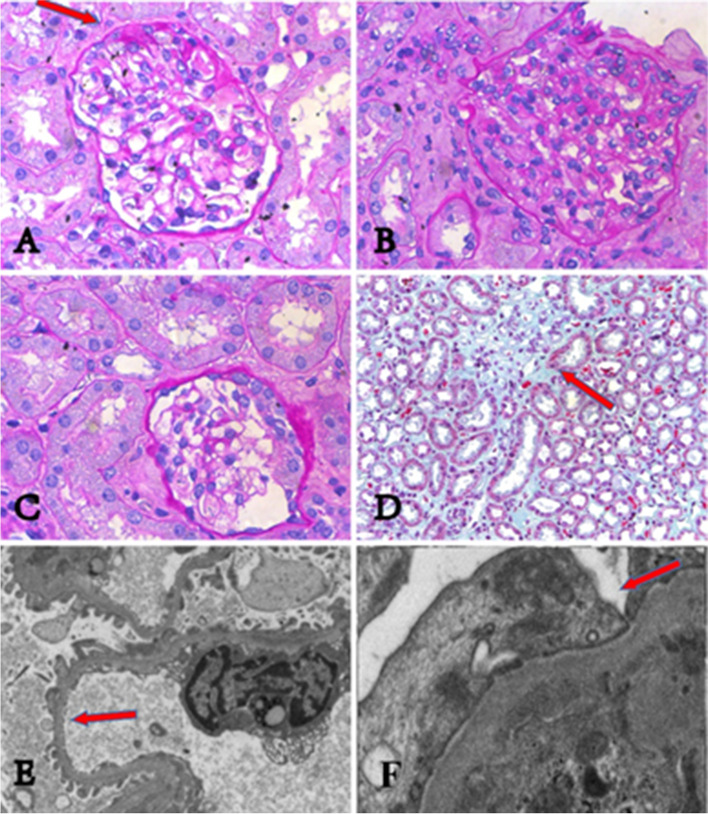
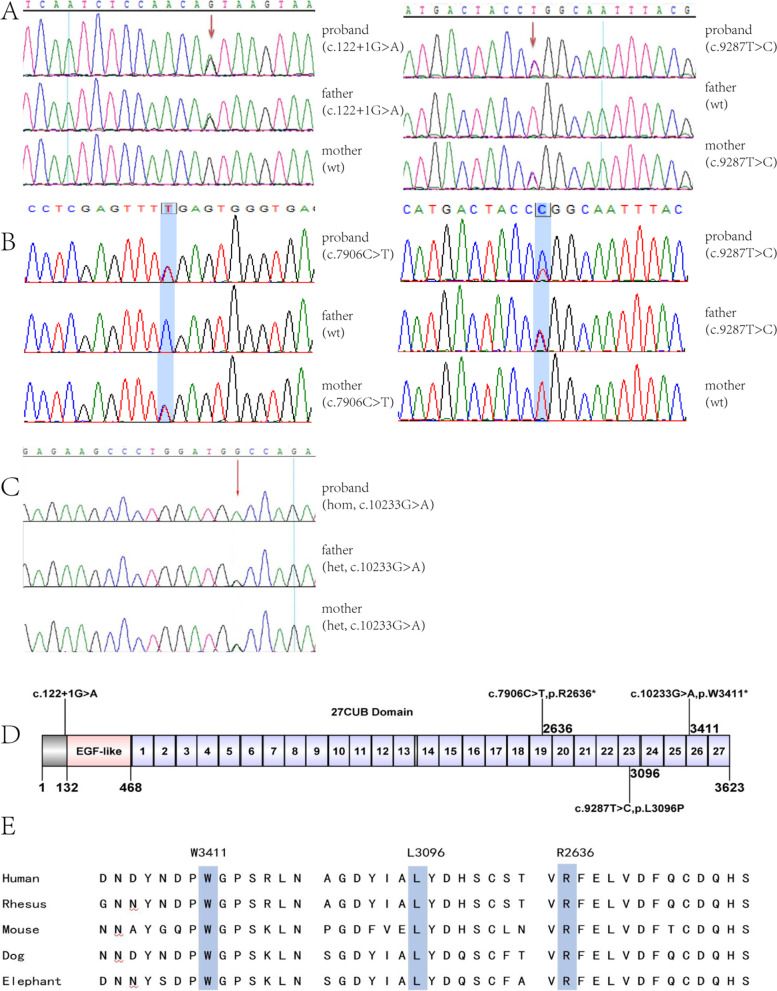
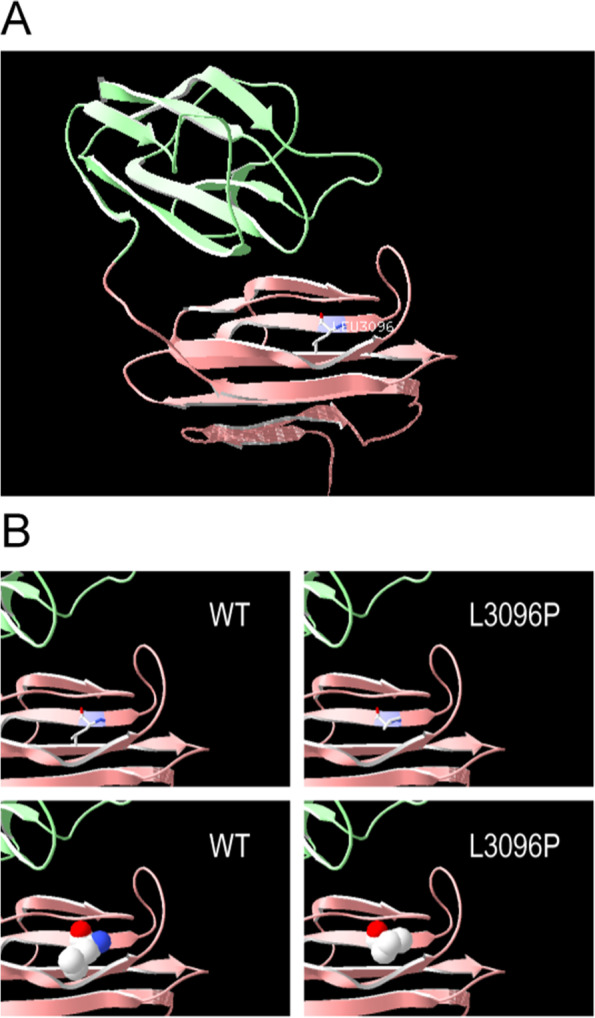
Results: All three children presented with isolated proteinuria, no megaloblastic anemia. Their urine levels of β2 microglobulin were normal or slightly higher. Renal biopsies showed focal segmental glomerulosclerosis with mild glomerular mesangial hypercellularity, partial effacement of foot processes and podocyte microvillation. Two of them were found to carry compound heterozygous mutations and one homozygous mutation of CUBN gene. Totally four CUBN gene biallelic pathogenic variants were identified, including c.9287 T > C (p.L3096P), c.122 + 1G > A, c.7906C > T (p.R2636*), c.10233G > A (p.W3411*). Except for intron splice-site mutation, all other variants are located in highly conserved sites of CUB domain for binding to albumin.
Conclusion: The results demonstrate that CUBN gene mutations may cause isolated proteinuria pathologically presented as FSGS. Our cases extend the spectrum of renal manifestation and genotype of CUBN gene mutations.
Keywords: CUBN gene; Focal segmental glomerulosclerosis; Gene mutation; Podocyte; Proteinuria.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
