

Fast nanopore sequencing data analysis with SLOW5
- PMID: 34980914
- PMCID: PMC9287168
- DOI: 10.1038/s41587-021-01147-4
Fast nanopore sequencing data analysis with SLOW5
Abstract
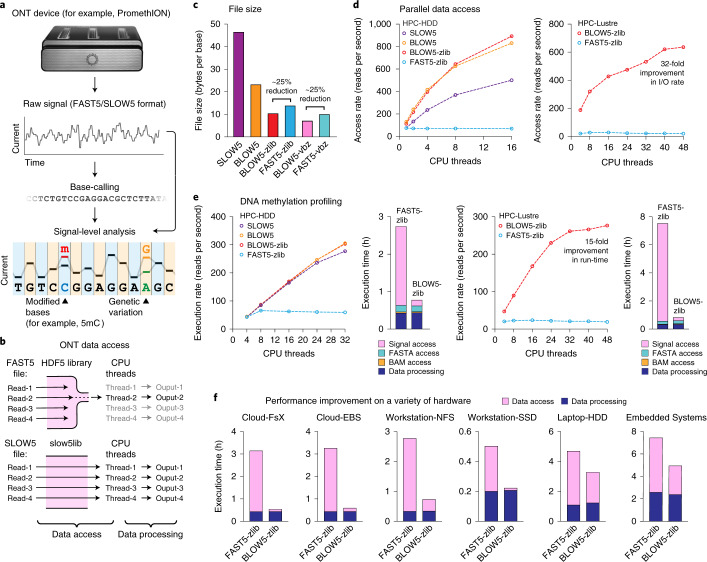
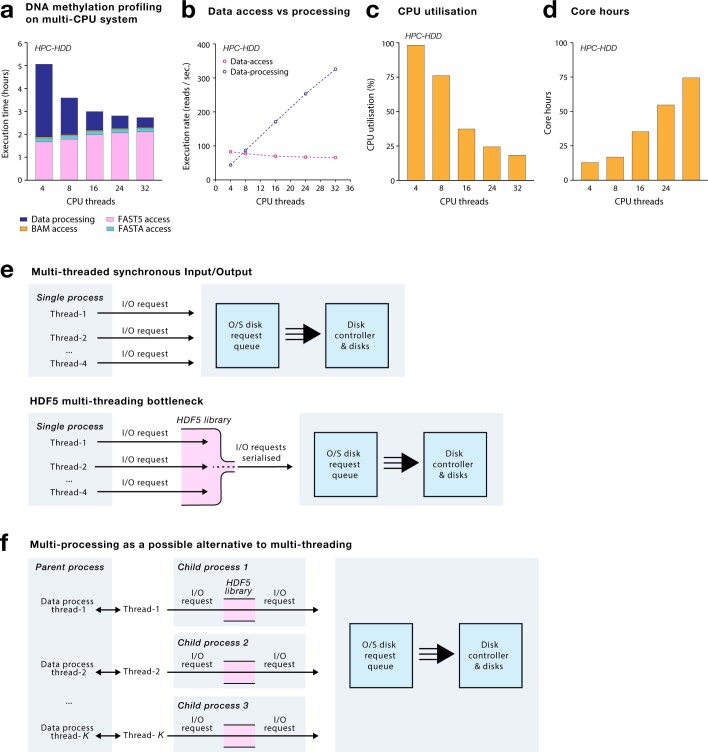
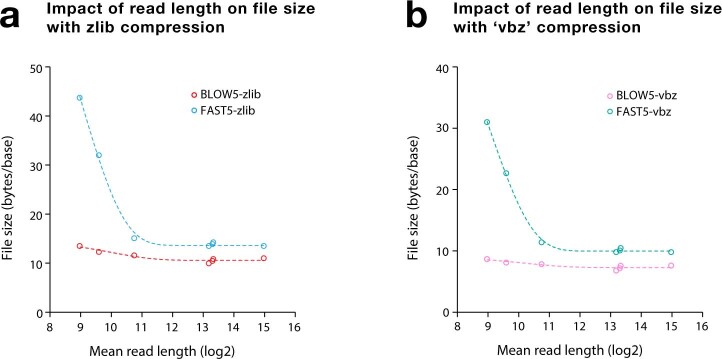
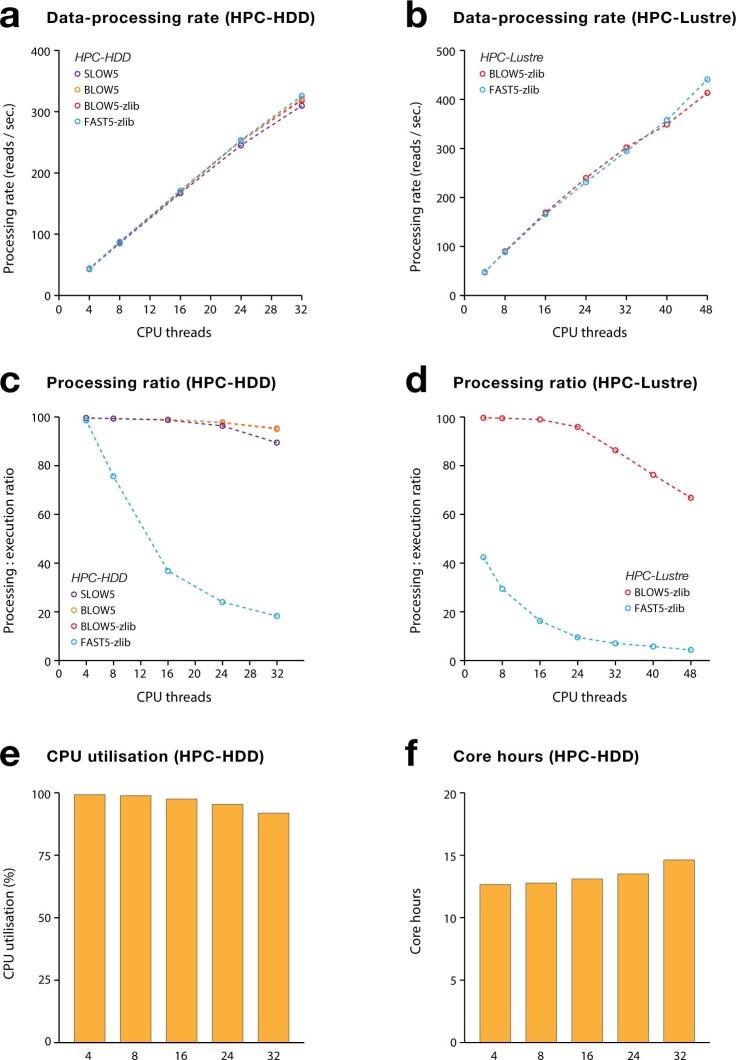
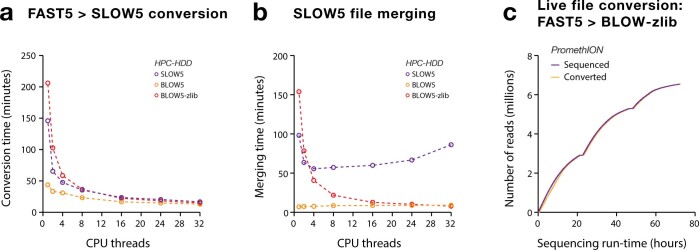
Nanopore sequencing depends on the FAST5 file format, which does not allow efficient parallel analysis. Here we introduce SLOW5, an alternative format engineered for efficient parallelization and acceleration of nanopore data analysis. Using the example of DNA methylation profiling of a human genome, analysis runtime is reduced from more than two weeks to approximately 10.5 h on a typical high-performance computer. SLOW5 is approximately 25% smaller than FAST5 and delivers consistent improvements on different computer architectures.
© 2022. Crown.
Conflict of interest statement
I.W.D. manages a fee-for-service nanopore sequencing facility at the Garvan Institute of Medical Research, which is a customer of Oxford Nanopore Technologies but has no further financial relationship. H.G., H. Samarakoon, J.M.F., J.M.H. and M.A.S. have received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. The authors declare no other competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
