

Genetic variation influencing DNA methylation provides insights into molecular mechanisms regulating genomic function
- PMID: 34980917
- PMCID: PMC7617265
- DOI: 10.1038/s41588-021-00969-x
Genetic variation influencing DNA methylation provides insights into molecular mechanisms regulating genomic function
Abstract
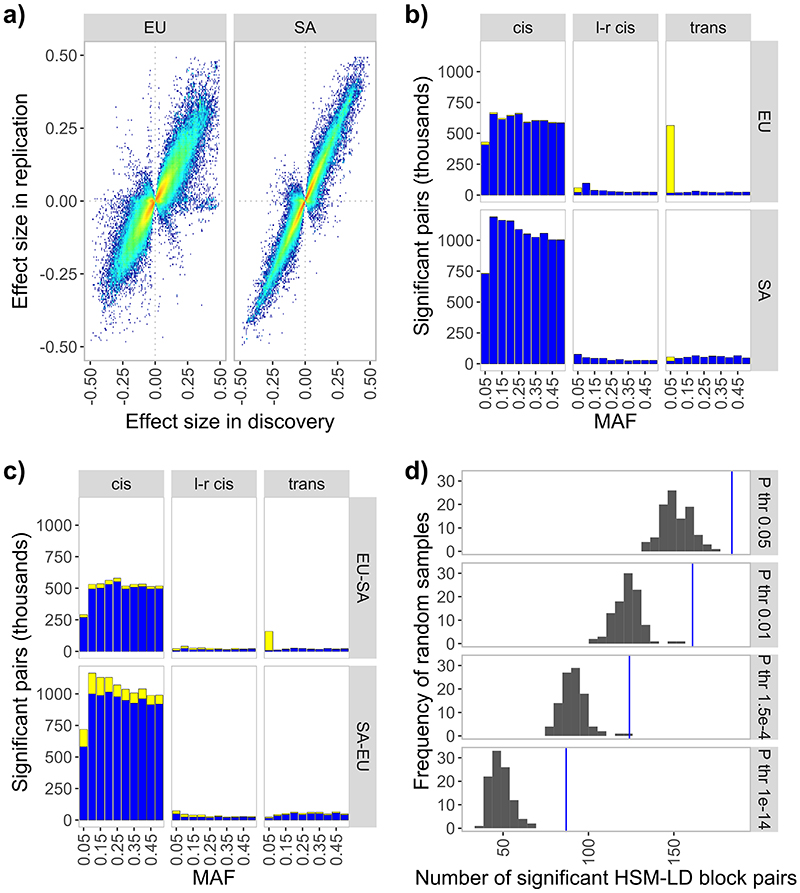
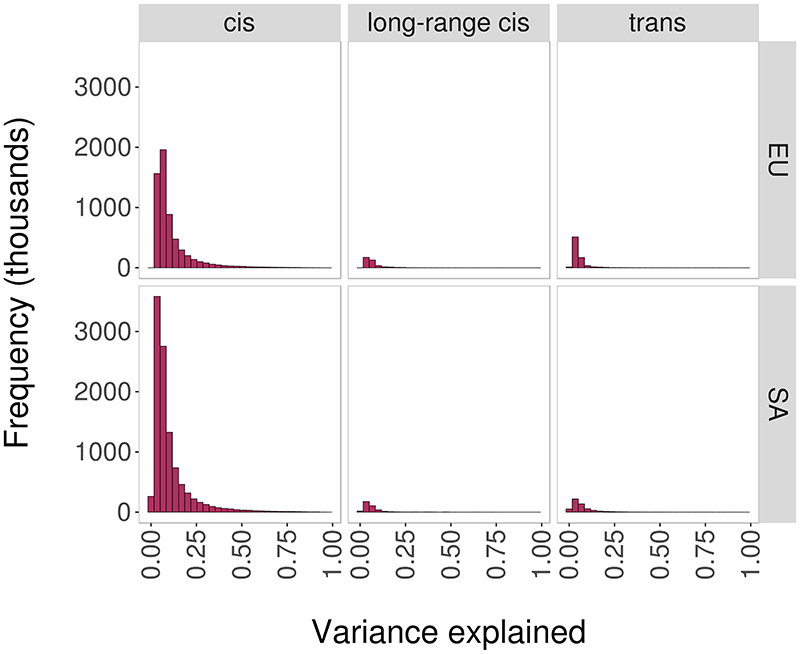
We determined the relationships between DNA sequence variation and DNA methylation using blood samples from 3,799 Europeans and 3,195 South Asians. We identify 11,165,559 SNP-CpG associations (methylation quantitative trait loci (meQTL), P < 10-14), including 467,915 meQTL that operate in trans. The meQTL are enriched for functionally relevant characteristics, including shared chromatin state, High-throuhgput chromosome conformation interaction, and association with gene expression, metabolic variation and clinical traits. We use molecular interaction and colocalization analyses to identify multiple nuclear regulatory pathways linking meQTL loci to phenotypic variation, including UBASH3B (body mass index), NFKBIE (rheumatoid arthritis), MGA (blood pressure) and COMMD7 (white cell counts). For rs6511961 , chromatin immunoprecipitation followed by sequencing (ChIP-seq) validates zinc finger protein (ZNF)333 as the likely trans acting effector protein. Finally, we used interaction analyses to identify population- and lineage-specific meQTL, including rs174548 in FADS1, with the strongest effect in CD8+ T cells, thus linking fatty acid metabolism with immune dysregulation and asthma. Our study advances understanding of the potential pathways linking genetic variation to human phenotype.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures














References
-
- Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. - PubMed
-
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–6. - PubMed
-
- Parry A, Rulands S, Reik W. Active turnover of DNA methylation during cell fate decisions. Nat Rev Genet. 2021;22:59–66. - PubMed
-
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. - PubMed
Publication types
MeSH terms
Grants and funding
- G0601966/MRC_/Medical Research Council/United Kingdom
- R01 MH063706/MH/NIMH NIH HHS/United States
- SP/04/002/BHF_/British Heart Foundation/United Kingdom
- MC_UP_1605/7/MRC_/Medical Research Council/United Kingdom
- 081917/WT_/Wellcome Trust/United Kingdom
- RG/14/5/30893/BHF_/British Heart Foundation/United Kingdom
- BB/S020845/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 081917/Z/07/Z/WT_/Wellcome Trust/United Kingdom
- G0500539/MRC_/Medical Research Council/United Kingdom
- BB/J004480/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- RG/14/5/30893/BHF_/British Heart Foundation/United Kingdom
- RL1 MH083268/MH/NIMH NIH HHS/United States
- 084723/Z/08/Z/WT_/Wellcome Trust/United Kingdom
- G1002319/MRC_/Medical Research Council/United Kingdom
- R01 HL087679/HL/NHLBI NIH HHS/United States
- 212945/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- 084723/WT_/Wellcome Trust/United Kingdom
- G0600705/MRC_/Medical Research Council/United Kingdom
- MR/L007150/1/MRC_/Medical Research Council/United Kingdom
- G0700931/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
