

Cellular and molecular basis of thyroid autoimmunity
- PMID: 34981746
- PMCID: PMC9142813
- DOI: 10.1530/ETJ-21-0024
Cellular and molecular basis of thyroid autoimmunity
Abstract
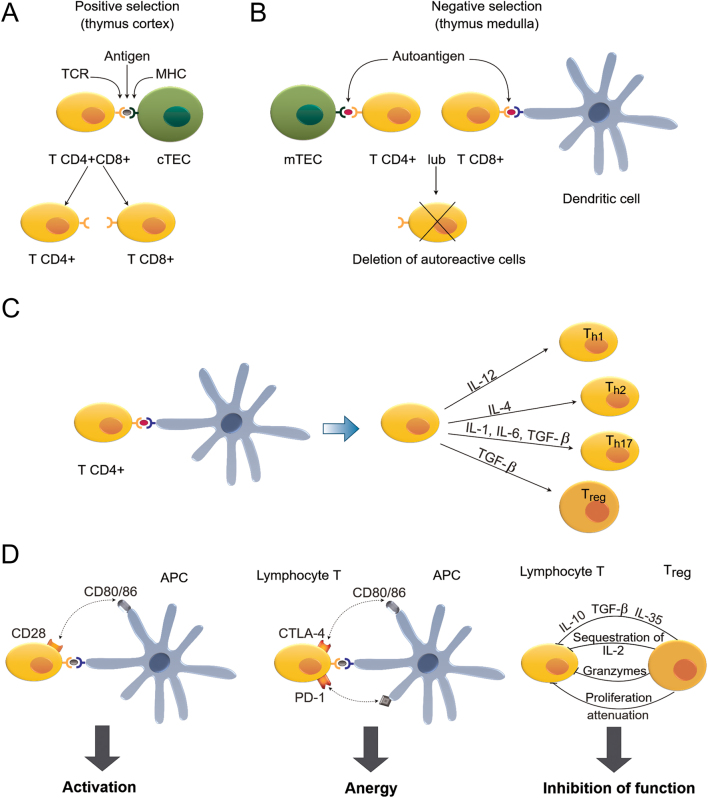
Autoimmune thyroid disease (AITD) is the most common human autoimmune disease. The two major clinical manifestations of AITD are Graves' disease and Hashimoto's thyroiditis (HT). AITD is characterized by lymphocytic infiltration of the thyroid gland, leading either to follicular cell damage, thyroid gland destruction, and development of hypothyroidism (in HT) or thyroid hyperplasia, induced by thyroid antibodies which activate thyrotropin receptor (TSHR) on thyrocytes, leading to hyperthyroidism. The aim of this review is to present up-to-date picture of the molecular and cellular mechanisms that underlie the pathology of AITD. Based on studies involving patients, animal AITD models, and thyroid cell lines, we discuss the key events leading to the loss of immune tolerance to thyroid autoantigens as well as the signaling cascades leading to the destruction of thyroid gland. Special focus is given on the interplay between the environmental and genetic factors, as well as ncRNAs and microbiome contributing to AITD development. In particular, we describe mechanistic models by which SNPs in genes involved in immune regulation and thyroid function, such as CD40, TSHR, FLT3, and PTPN22, underlie AITD predisposition. The clinical significance of novel diagnostic and prognostic biomarkers based on ncRNAs and microbiome composition is also underscored. Finally, we discuss the possible significance of probiotic supplementation on thyroid function in AITD.
Keywords: AITD; Graves’ disease; Hashimoto’s thyroiditis; autoimmune thyroid disease; circRNAs; lncRNAs; miRNAs; microbiome; non-coding RNAs; thyroid antigens; thyroid autoimmunity.
Figures







References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
