

Addressing dyslipidemic risk beyond LDL-cholesterol
- PMID: 34981790
- PMCID: PMC8718149
- DOI: 10.1172/JCI148559
Addressing dyslipidemic risk beyond LDL-cholesterol
Abstract
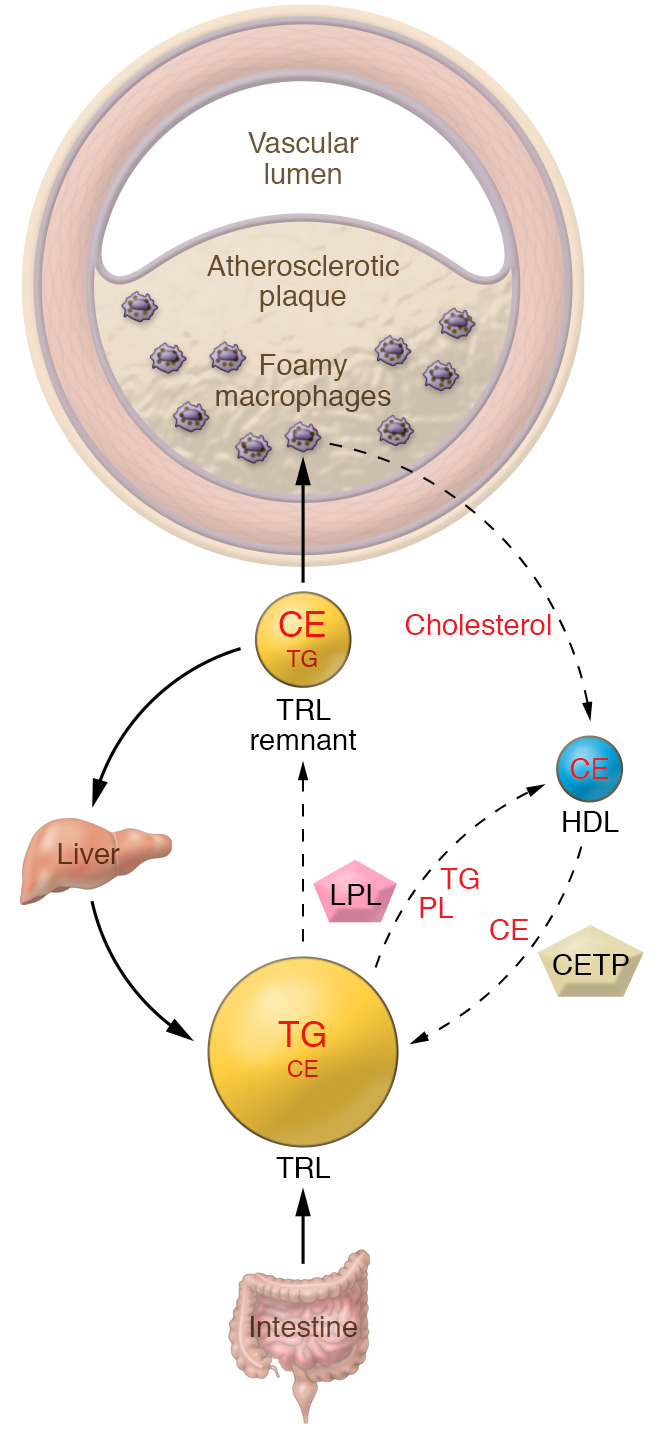
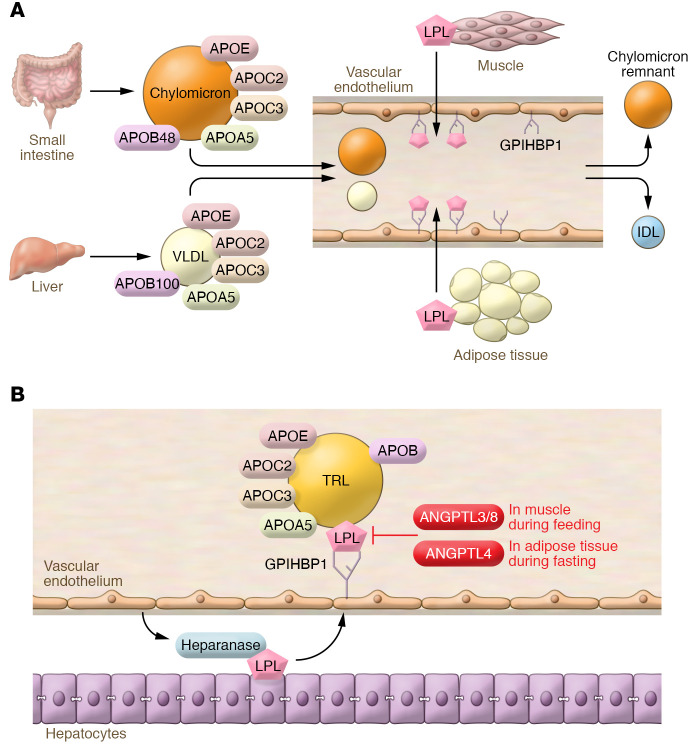
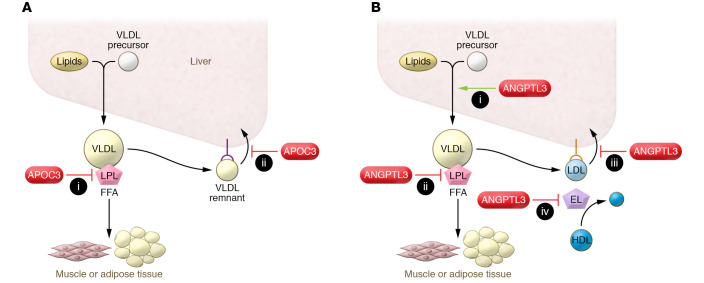
Despite the success of LDL-lowering drugs in reducing cardiovascular disease (CVD), there remains a large burden of residual disease due in part to persistent dyslipidemia characterized by elevated levels of triglyceride-rich lipoproteins (TRLs) and reduced levels of HDL. This form of dyslipidemia is increasing globally as a result of the rising prevalence of obesity and metabolic syndrome. Accumulating evidence suggests that impaired hepatic clearance of cholesterol-rich TRL remnants leads to their accumulation in arteries, promoting foam cell formation and inflammation. Low levels of HDL may associate with reduced cholesterol efflux from foam cells, aggravating atherosclerosis. While fibrates and fish oils reduce TRL, they have not been uniformly successful in reducing CVD, and there is a large unmet need for new approaches to reduce remnants and CVD. Rare genetic variants that lower triglyceride levels via activation of lipolysis and associate with reduced CVD suggest new approaches to treating dyslipidemia. Apolipoprotein C3 (APOC3) and angiopoietin-like 3 (ANGPTL3) have emerged as targets for inhibition by antibody, antisense, or RNAi approaches. Inhibition of either molecule lowers TRL but respectively raises or lowers HDL levels. Large clinical trials of such agents in patients with high CVD risk and elevated levels of TRL will be required to demonstrate efficacy of these approaches.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
