

Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease
- PMID: 34983592
- PMCID: PMC8729103
- DOI: 10.1186/s12974-021-02354-1
Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease
Abstract
Patients with inflammatory bowel disease (IBD) suffer from depression at higher rates than the general population. An etiological trigger of depressive symptoms is theorised to be inflammation within the central nervous system. It is believed that heightened intestinal inflammation and dysfunction of the enteric nervous system (ENS) contribute to impaired intestinal permeability, which facilitates the translocation of intestinal enterotoxins into the blood circulation. Consequently, these may compromise the immunological and physiological functioning of distant non-intestinal tissues such as the brain. In vivo models of colitis provide evidence of increased blood-brain barrier permeability and enhanced central nervous system (CNS) immune activity triggered by intestinal enterotoxins and blood-borne inflammatory mediators. Understanding the immunological, physiological, and structural changes associated with IBD and neuroinflammation may aid in the development of more tailored and suitable pharmaceutical treatment for IBD-associated depression.
Keywords: Depression; Gut-brain axis; Inflammatory bowel disease; Neuroinflammation.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
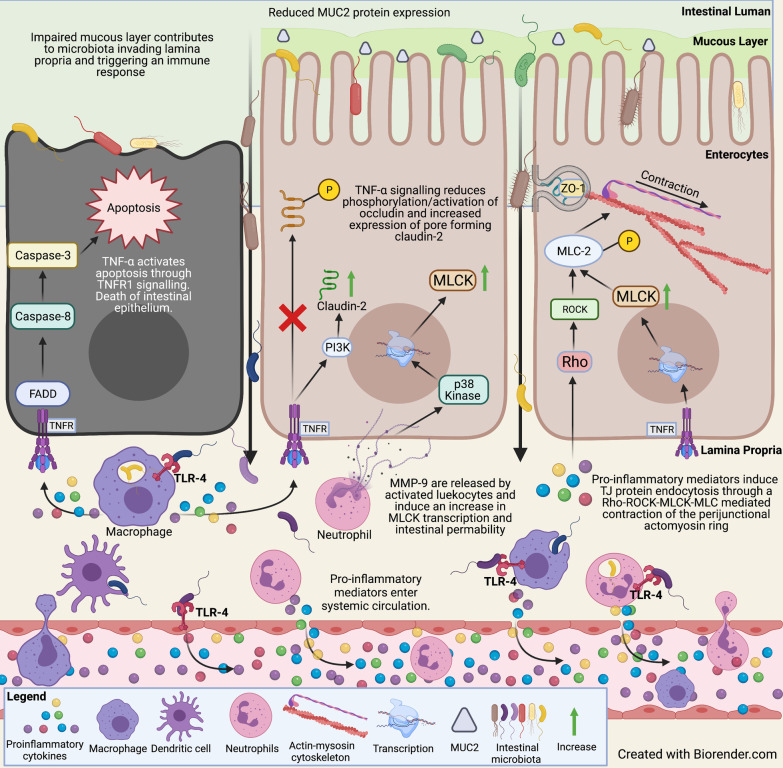
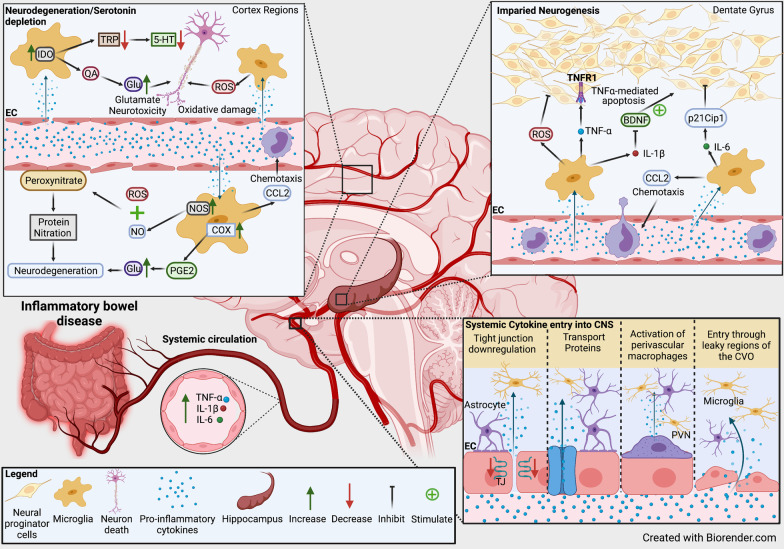
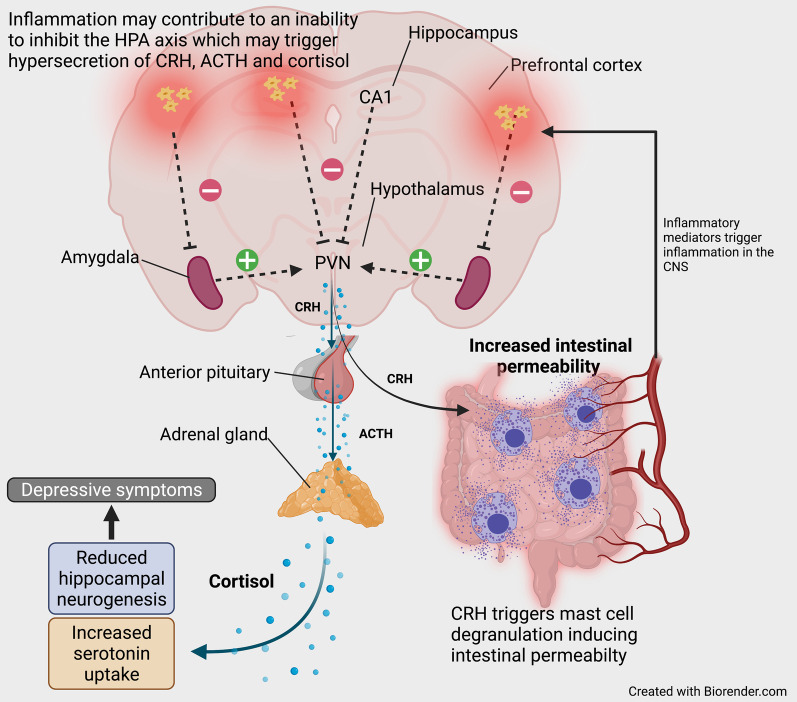
References
-
- Alatab S, Sepanlou SG, Ikuta K, Vahedi H, Bisignano C, Safiri S, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(1):17–30. - PMC - PubMed
-
- Nigro G, Angelini G, Grosso SB, Caula G, Sategna-Guidetti C. Psychiatric predictors of noncompliance in inflammatory bowel disease: psychiatry and compliance. J Clin Gastroenterol. 2001;32(1):1. - PubMed
-
- Gracie DJ, Hamlin PJ, Ford AC. The influence of the brain–gut axis in inflammatory bowel disease and possible implications for treatment. Lancet Gastroenterol Hepatol. 2019;4(8):632–642. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
