

Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders
- PMID: 34983696
- PMCID: PMC8725368
- DOI: 10.1186/s42466-021-00162-9
Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders
Abstract
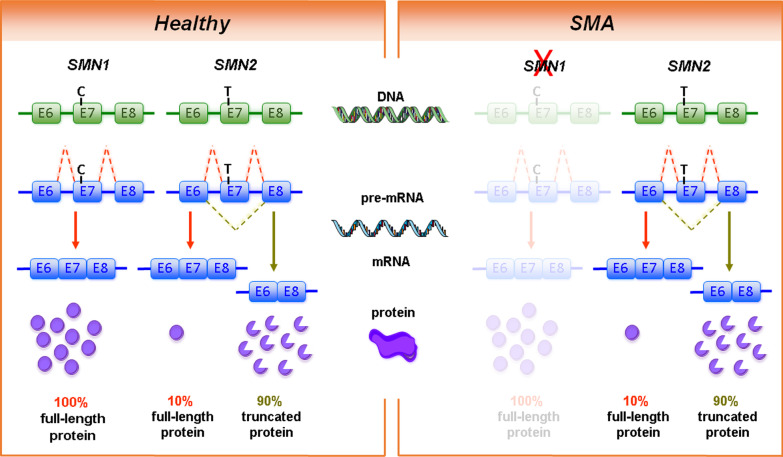
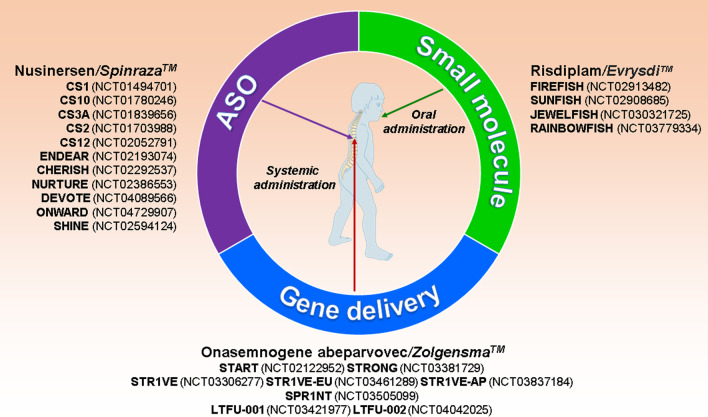
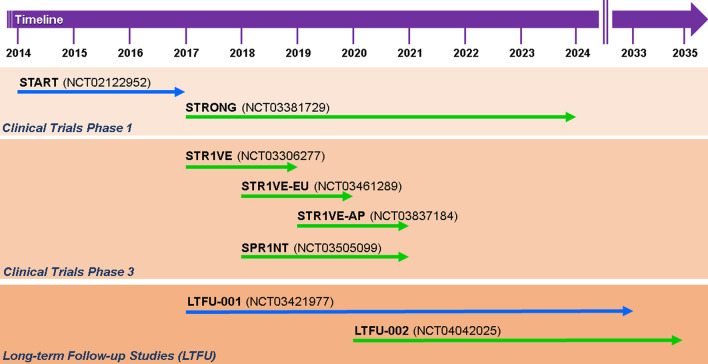
Background: Major efforts have been made in the last decade to develop and improve therapies for proximal spinal muscular atrophy (SMA). The introduction of Nusinersen/Spinraza™ as an antisense oligonucleotide therapy, Onasemnogene abeparvovec/Zolgensma™ as an AAV9-based gene therapy and Risdiplam/Evrysdi™ as a small molecule modifier of pre-mRNA splicing have set new standards for interference with neurodegeneration.
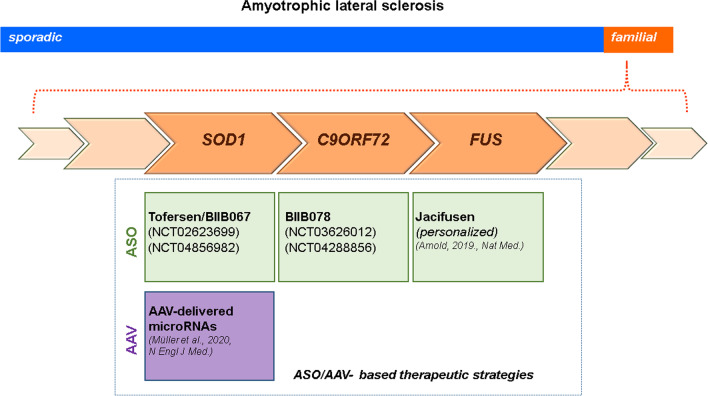
Main body: Therapies for SMA are designed to interfere with the cellular basis of the disease by modifying pre-mRNA splicing and enhancing expression of the Survival Motor Neuron (SMN) protein, which is only expressed at low levels in this disorder. The corresponding strategies also can be applied to other disease mechanisms caused by loss of function or toxic gain of function mutations. The development of therapies for SMA was based on the use of cell culture systems and mouse models, as well as innovative clinical trials that included readouts that had originally been introduced and optimized in preclinical studies. This is summarized in the first part of this review. The second part discusses current developments and perspectives for amyotrophic lateral sclerosis, muscular dystrophies, Parkinson's and Alzheimer's disease, as well as the obstacles that need to be overcome to introduce RNA-based therapies and gene therapies for these disorders.
Conclusion: RNA-based therapies offer chances for therapy development of complex neurodegenerative disorders such as amyotrophic lateral sclerosis, muscular dystrophies, Parkinson's and Alzheimer's disease. The experiences made with these new drugs for SMA, and also the experiences in AAV gene therapies could help to broaden the spectrum of current approaches to interfere with pathophysiological mechanisms in neurodegeneration.
Keywords: Alzheimer disease; Amyotrophic lateral sclerosis; Clinical trial; Gene therapy; Motoneuron disease; Muscular disease; Muscular dystrophy; Neurodegenerative disease; Parkinson disease; Spinal muscular atrophy.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Aartsma-Rus A, Straub V, Hemmings R, Haas M, Schlosser-Weber G, Stoyanova-Beninska V, Mercuri E, Muntoni F, Sepodes B, Vroom E, Balabanov P. Development of exon skipping therapies for duchenne muscular dystrophy: A critical review and a perspective on the outstanding issues [Review] Nucleic Acid Therapeutics. 2017;27(5):251–259. doi: 10.1089/nat.2017.0682. - DOI - PMC - PubMed
-
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Neuron. 2014;81(3):536–543. doi: 10.1016/j.neuron.2013.12.018. - DOI - PMC - PubMed
-
- Alrafiah A, Karyka E, Coldicott I, Iremonger K, Lewis KE, Ning K, Azzouz M. Plastin 3 promotes motor neuron axonal growth and extends survival in a mouse model of spinal muscular atrophy. Molecular Therapy-Methods & Clinical Development. 2018;9:81–89. doi: 10.1016/j.omtm.2018.01.007. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources