

A test of the pioneer factor hypothesis using ectopic liver gene activation
- PMID: 34984978
- PMCID: PMC8849321
- DOI: 10.7554/eLife.73358
A test of the pioneer factor hypothesis using ectopic liver gene activation
Abstract
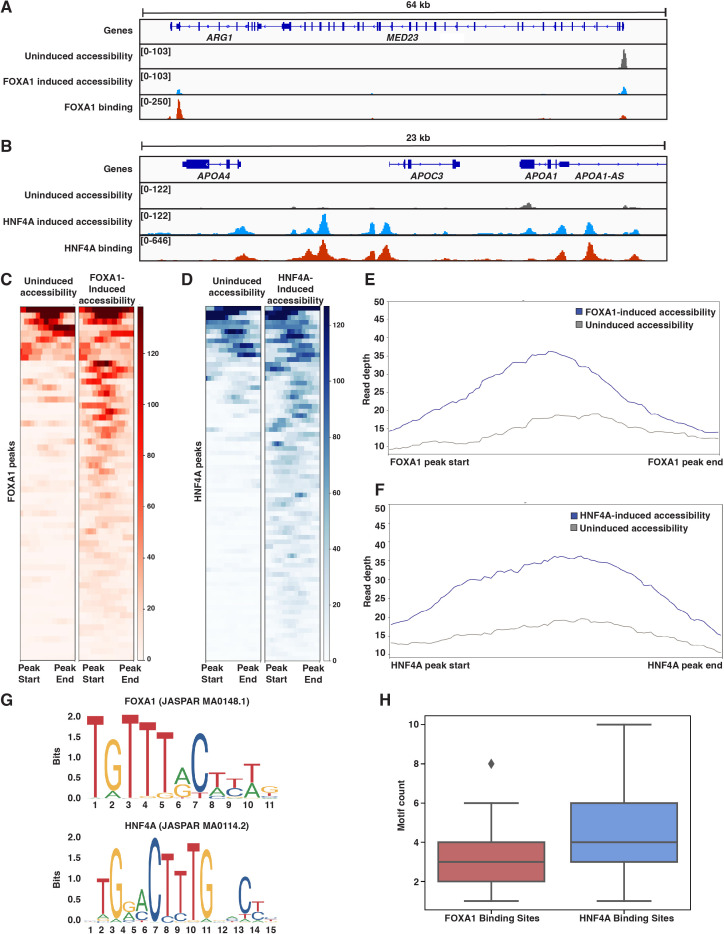
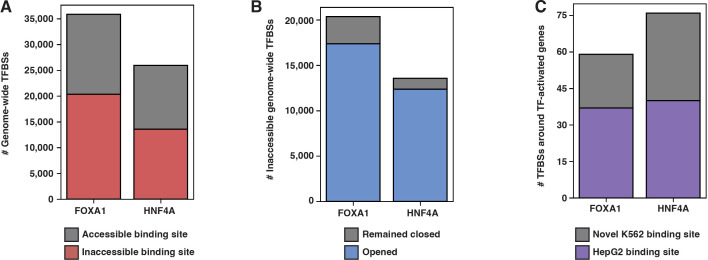
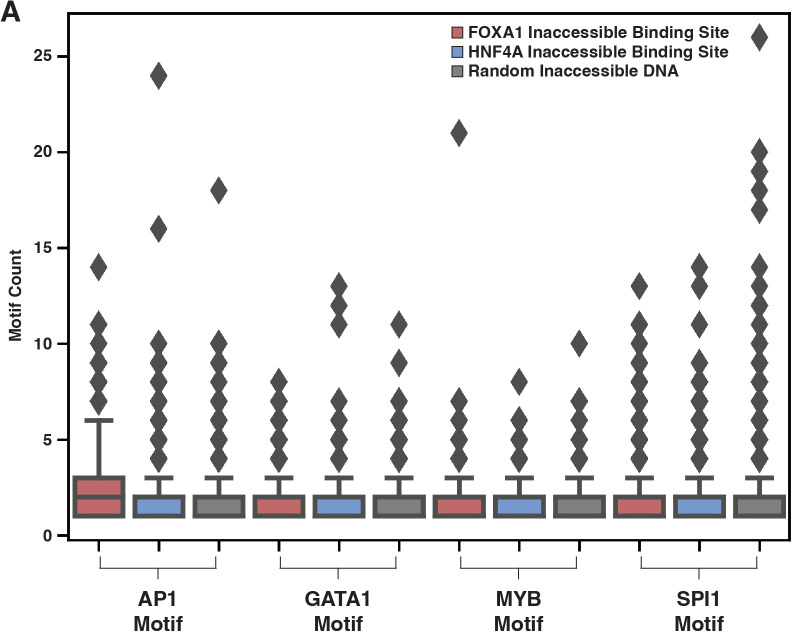
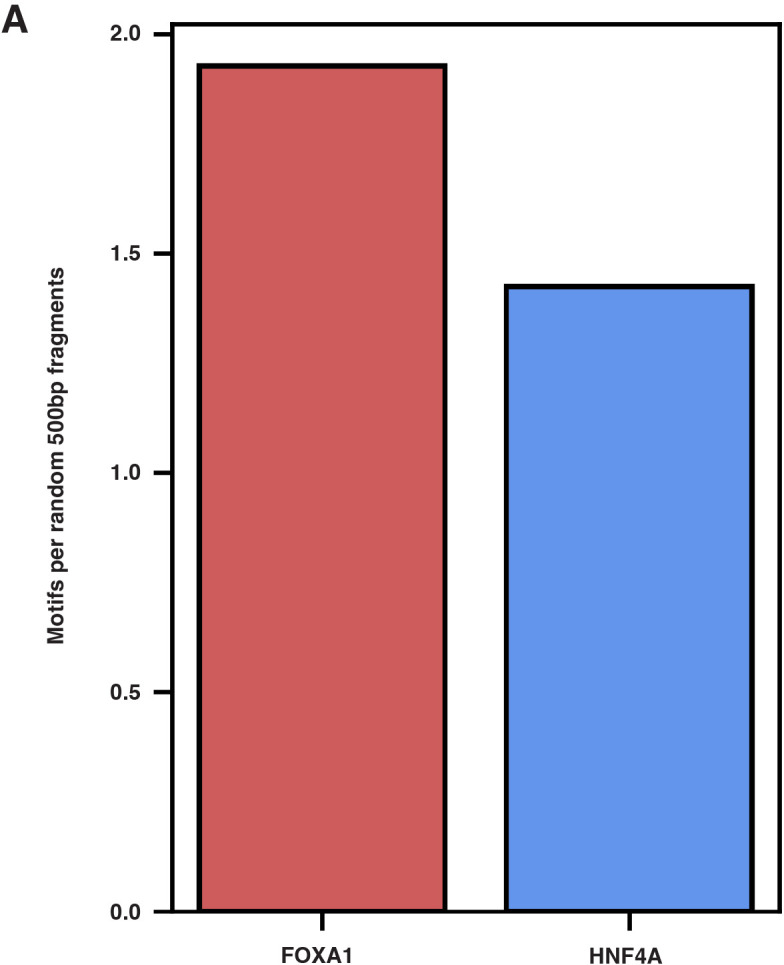
The pioneer factor hypothesis (PFH) states that pioneer factors (PFs) are a subclass of transcription factors (TFs) that bind to and open inaccessible sites and then recruit non-pioneer factors (non-PFs) that activate batteries of silent genes. The PFH predicts that ectopic gene activation requires the sequential activity of qualitatively different TFs. We tested the PFH by expressing the endodermal PF FOXA1 and non-PF HNF4A in K562 lymphoblast cells. While co-expression of FOXA1 and HNF4A activated a burst of endoderm-specific gene expression, we found no evidence for a functional distinction between these two TFs. When expressed independently, both TFs bound and opened inaccessible sites, activated endodermal genes, and 'pioneered' for each other, although FOXA1 required fewer copies of its motif for binding. A subset of targets required both TFs, but the predominant mode of action at these targets did not conform to the sequential activity predicted by the PFH. From these results, we hypothesize an alternative to the PFH where 'pioneer activity' depends not on categorically different TFs but rather on the affinity of interaction between TF and DNA.
Keywords: cellular reprogramming; chromosomes; gene expression; genetics; genomics; human k562 cell line; pioneer factors; transcription factors.
Plain language summary
Cells only use a fraction of their genetic information to make the proteins they need. The rest is carefully packaged away and tightly bundled in structures called nucleosomes. This physically shields the DNA from being accessed by transcription factors – the molecular actors that can read genes and kickstart the protein production process. Effectively, the genetic sequences inside nucleosomes are being silenced. However, during development, transcription factors must overcome this nucleosome barrier and activate silent genes to program cells. The pioneer factor hypothesis describes how this may be possible: first, ‘pioneer’ transcription factors can bind to and ‘open up’ nucleosomes to make target genes accessible. Then, non-pioneer factors can access the genetic sequence and recruit cofactors that begin copying the now-exposed genetic information. The widely accepted theory is based on studies of two proteins – FOXA1, an archetypal pioneer factor, and HNF4A, a non-pioneer factor – but the predictions of the pioneer factor hypothesis have yet to be explicitly tested. To do so, Hansen et al. expressed FOXA1 and HNF4A, separately and together, in cells which do not usually make these proteins. They then assessed how the proteins could bind to DNA and impact gene accessibility and transcription. The experiments demonstrate that FOXA1 and HNF4A do not necessarily follow the two-step activation predicted by the pioneer factor hypothesis. When expressed independently, both transcription factors bound and opened inaccessible sites, activated target genes, and ‘pioneered’ for each other. Similar patterns were observed across the genome. The only notable distinction between the two factors was that FOXA1, the archetypal pioneering factor, required fewer copies of its target sequence to bind DNA than HNF4A. These findings led Hansen et al. to propose an alternative theory to the pioneer factor hypothesis which eliminates the categorical distinction between pioneer and non-pioneer factors. Overall, this work has implications for how biologists understand the way that transcription factors activate silent genes during development.
© 2022, Hansen et al.
Conflict of interest statement
JH, KL, BC No competing interests declared
Figures
Similar articles
-
A quantitative metric of pioneer activity reveals that HNF4A has stronger in vivo pioneer activity than FOXA1.Genome Biol. 2022 Oct 17;23(1):221. doi: 10.1186/s13059-022-02792-x. Genome Biol. 2022. PMID: 36253868 Free PMC article.
-
Different chromatin-scanning modes lead to targeting of compacted chromatin by pioneer factors FOXA1 and SOX2.Cell Rep. 2023 Jul 25;42(7):112748. doi: 10.1016/j.celrep.2023.112748. Epub 2023 Jul 4. Cell Rep. 2023. PMID: 37405916 Free PMC article.
-
Redirecting the pioneering function of FOXA1 with covalent small molecules.Mol Cell. 2024 Nov 7;84(21):4125-4141.e10. doi: 10.1016/j.molcel.2024.09.024. Epub 2024 Oct 15. Mol Cell. 2024. PMID: 39413792
-
The Role of FOXA1 in Human Normal Development and Its Functions in Sex Hormone-Related Cancers.Front Biosci (Landmark Ed). 2024 Jun 21;29(6):225. doi: 10.31083/j.fbl2906225. Front Biosci (Landmark Ed). 2024. PMID: 38940052 Review.
-
Pioneer of prostate cancer: past, present and the future of FOXA1.Protein Cell. 2021 Jan;12(1):29-38. doi: 10.1007/s13238-020-00786-8. Epub 2020 Sep 18. Protein Cell. 2021. PMID: 32946061 Free PMC article. Review.
Cited by
-
Time-varying stimuli that prolong IKK activation promote nuclear remodeling and mechanistic switching of NF-κB dynamics.bioRxiv [Preprint]. 2024 Oct 1:2024.09.26.615244. doi: 10.1101/2024.09.26.615244. bioRxiv. 2024. Update in: Nat Commun. 2025 Aug 8;16(1):7329. doi: 10.1038/s41467-025-62837-0. PMID: 39386677 Free PMC article. Updated. Preprint.
-
Structural dynamics in chromatin unraveling by pioneer transcription factors.Biophys Rev. 2024 Jul 4;16(3):365-382. doi: 10.1007/s12551-024-01205-6. eCollection 2024 Jun. Biophys Rev. 2024. PMID: 39099839 Free PMC article. Review.
-
The Drosophila embryo as a tabula rasa for the epigenome.Fac Rev. 2022 Dec 23;11:40. doi: 10.12703/r/11-40. eCollection 2022. Fac Rev. 2022. PMID: 36644296 Free PMC article. Review.
-
Pioneer factors - key regulators of chromatin and gene expression.Nat Rev Genet. 2023 Dec;24(12):809-815. doi: 10.1038/s41576-023-00648-z. Epub 2023 Sep 22. Nat Rev Genet. 2023. PMID: 37740118 No abstract available.
-
Downregulation of HNF4A enables transcriptomic reprogramming during the hepatic acute-phase response.Commun Biol. 2024 May 16;7(1):589. doi: 10.1038/s42003-024-06288-1. Commun Biol. 2024. PMID: 38755249 Free PMC article.
References
-
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung M-H, Trump S, Lightman SL, Vinson C, Stamatoyannopoulos JA, Hager GL. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Molecular Cell. 2011;43:145–155. doi: 10.1016/j.molcel.2011.06.016. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
