

Vasculopathy in COVID-19
- PMID: 34986238
- PMCID: PMC8736280
- DOI: 10.1182/blood.2021012250
Vasculopathy in COVID-19
Abstract
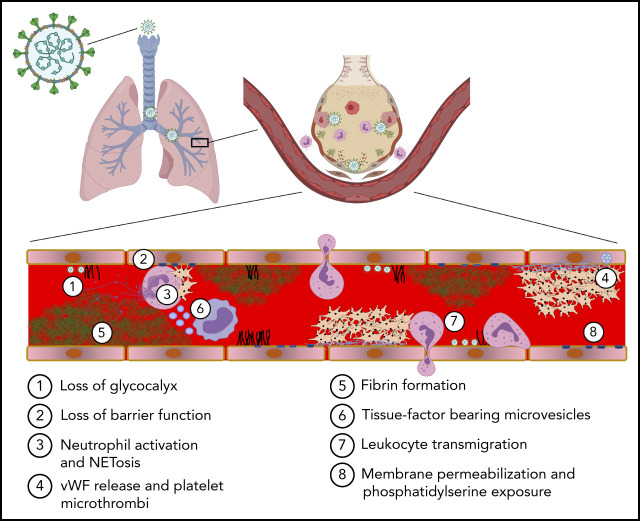
COVID-19 is a primary respiratory illness that is frequently complicated by systemic involvement of the vasculature. Vascular involvement leads to an array of complications ranging from thrombosis to pulmonary edema secondary to loss of barrier function. This review will address the vasculopathy of COVID-19 with a focus on the role of the endothelium in orchestrating the systemic response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. The endothelial receptor systems and molecular pathways activated in the setting of COVID-19 and the consequences of these inflammatory and prothrombotic changes on endothelial cell function will be discussed. The sequelae of COVID-19 vascular involvement at the level of organ systems will also be addressed, with an emphasis on the pulmonary vasculature but with consideration of effects on other vascular beds. The dramatic changes in endothelial phenotypes associated with COVID-19 has enabled the identification of biomarkers that could help guide therapy and predict outcomes. Knowledge of vascular pathogenesis in COVID-19 has also informed therapeutic approaches that may control its systemic sequelae. Because our understanding of vascular response in COVID-19 continues to evolve, we will consider areas of controversy, such as the extent to which SARS-CoV-2 directly infects endothelium and the degree to which vascular responses to SARS-CoV-2 are unique or common to those of other viruses capable of causing severe respiratory disease. This conceptual framework describing how SARS-CoV-2 infection affects endothelial inflammation, prothrombotic transformation, and barrier dysfunction will provide a context for interpreting new information as it arises addressing the vascular complications of COVID-19.
© 2022 by The American Society of Hematology.
Figures





References
-
- David S, Kümpers P, van Slyke P, Parikh SM. Mending leaky blood vessels: the angiopoietin-Tie2 pathway in sepsis. J Pharmacol Exp Ther. 2013;345(1):2-6. - PubMed
-
- Ferrario CM, Jessup J, Chappell MC, et al. . Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):2605-2610. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
