

miR-338-3p blocks TGFβ-induced myofibroblast differentiation through the induction of PTEN
- PMID: 34986654
- PMCID: PMC8884407
- DOI: 10.1152/ajplung.00251.2021
miR-338-3p blocks TGFβ-induced myofibroblast differentiation through the induction of PTEN
Abstract
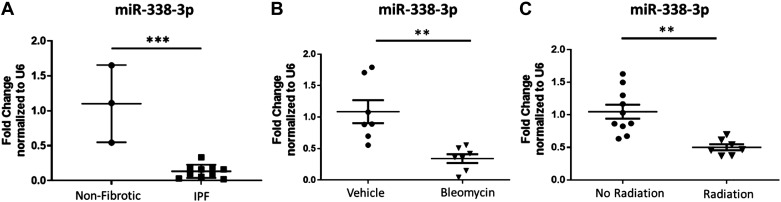
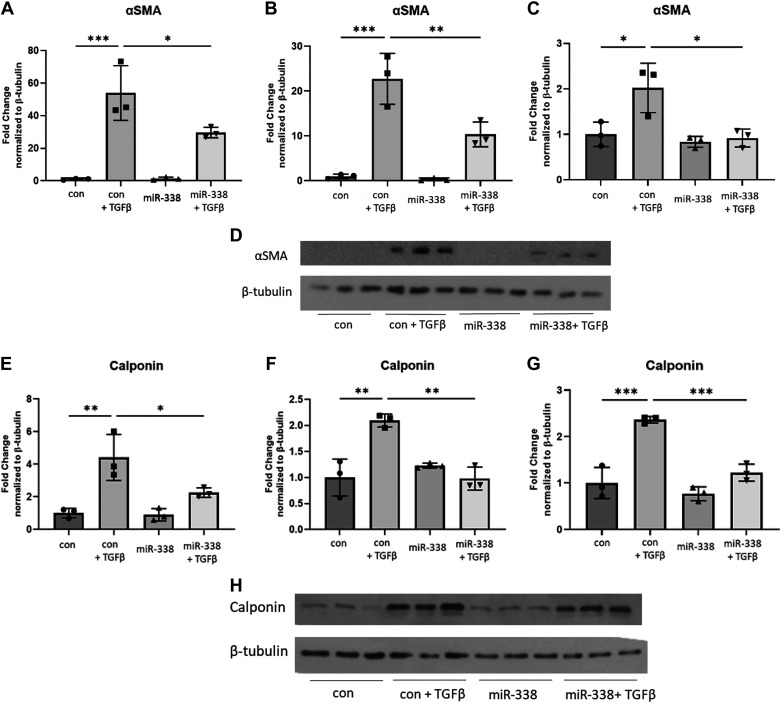
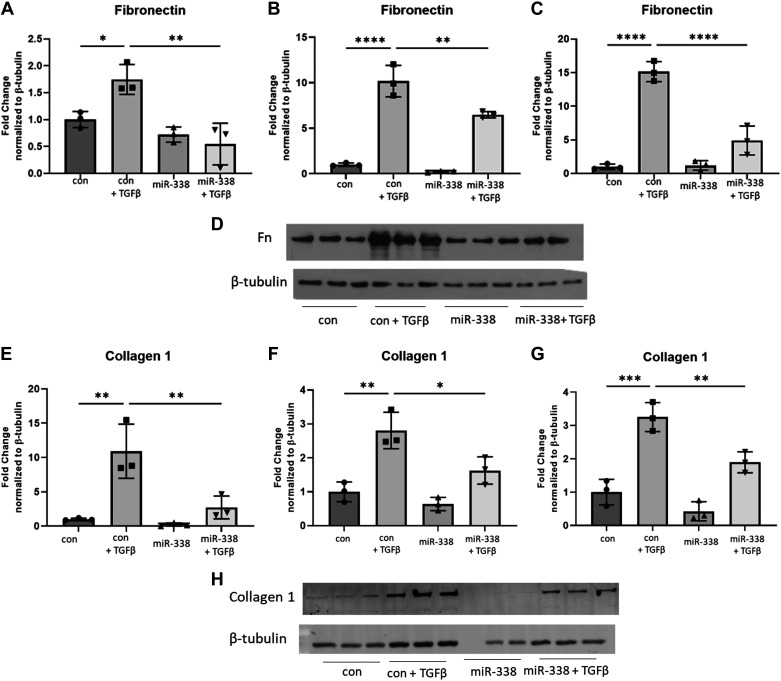
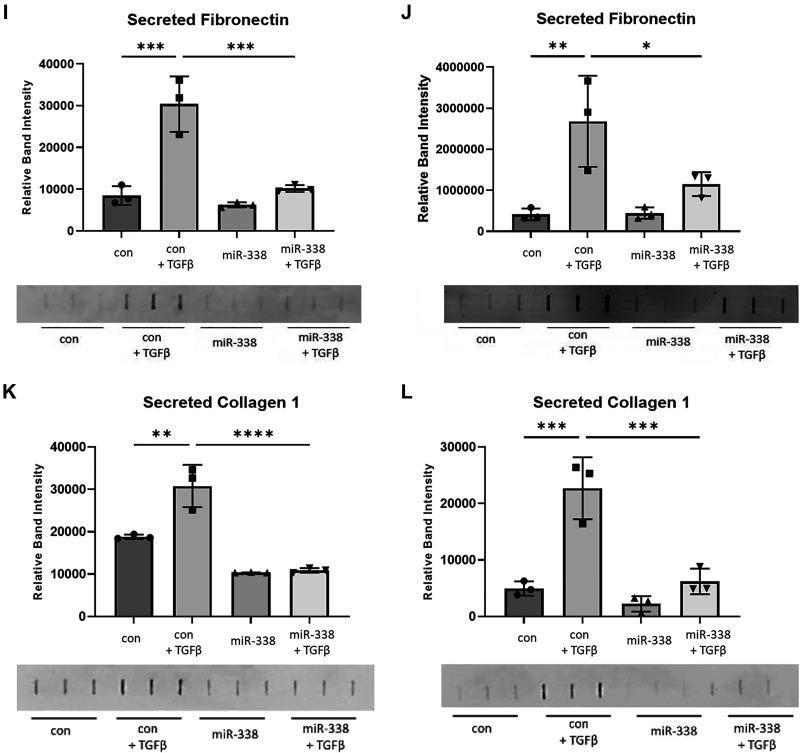
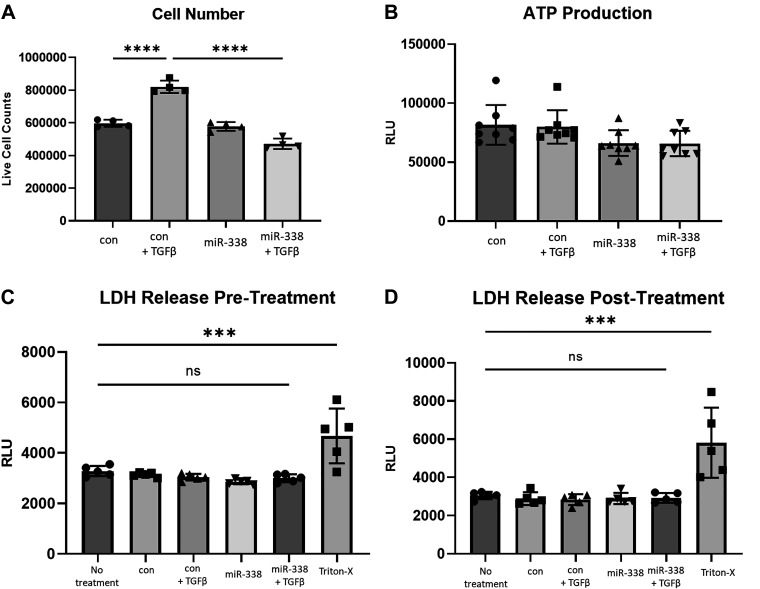
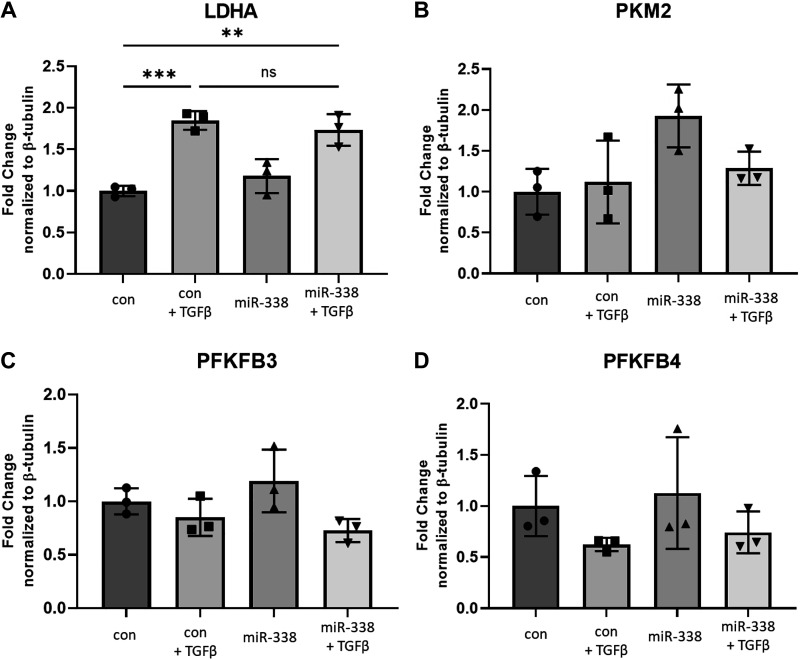
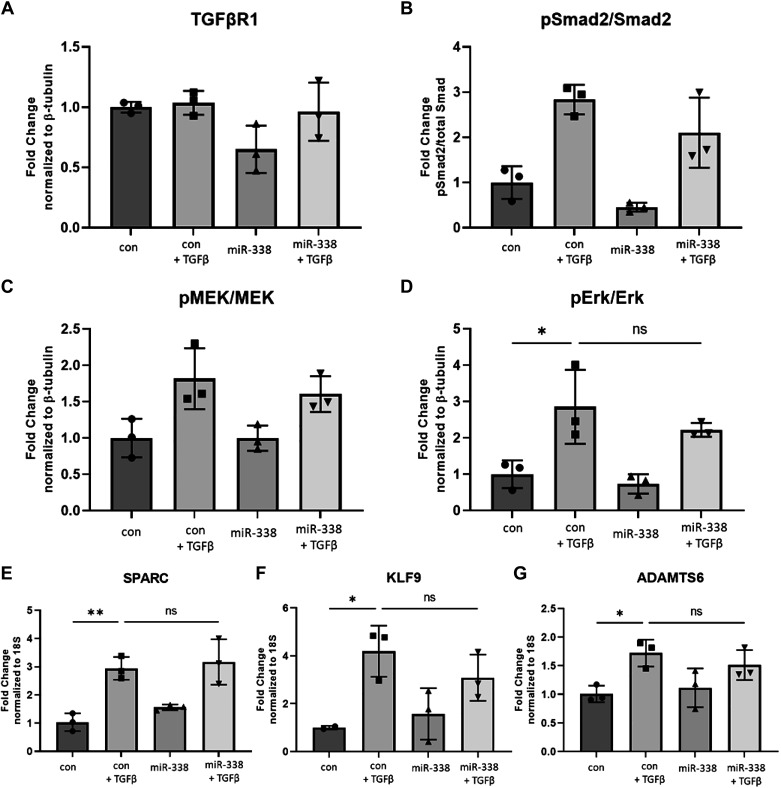
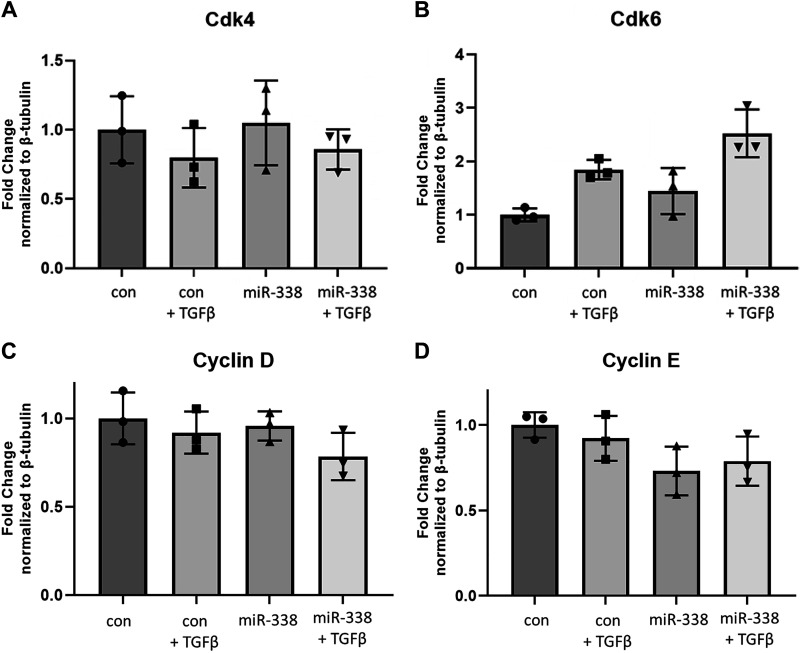
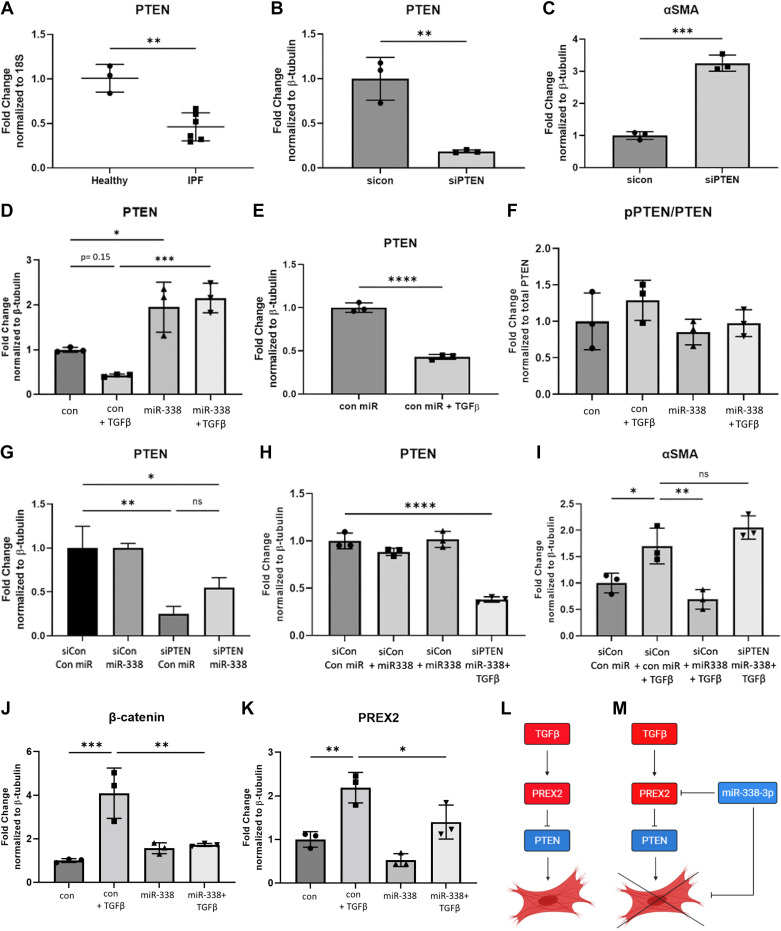
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease. The pathogenesis of IPF is not completely understood. However, numerous genes are associated with the development and progression of pulmonary fibrosis, indicating there is a significant genetic component to the pathogenesis of IPF. Epigenetic influences on the development of human disease, including pulmonary fibrosis, remain to be fully elucidated. In this paper, we identify miR-338-3p as a microRNA severely downregulated in the lungs of patients with pulmonary fibrosis and in experimental models of pulmonary fibrosis. Treatment of primary human lung fibroblasts with miR-338-3p inhibits myofibroblast differentiation and matrix protein production. Published and proposed targets of miR-338-3p such as TGFβ receptor 1, MEK/ERK 1/2, Cdk4, and Cyclin D are also not responsible for the regulation of pulmonary fibroblast behavior by miR-338-3p. miR-338-3p inhibits myofibroblast differentiation by preventing TGFβ-mediated downregulation of phosphatase and tensin homolog (PTEN), a known antifibrotic mediator.
Keywords: fibroblast; lung; miRNA; pulmonary fibrosis.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous