

Hemin-Induced Death Models Hemorrhagic Stroke and Is a Variant of Classical Neuronal Ferroptosis
- PMID: 34987108
- PMCID: PMC8916756
- DOI: 10.1523/JNEUROSCI.0923-20.2021
Hemin-Induced Death Models Hemorrhagic Stroke and Is a Variant of Classical Neuronal Ferroptosis
Abstract
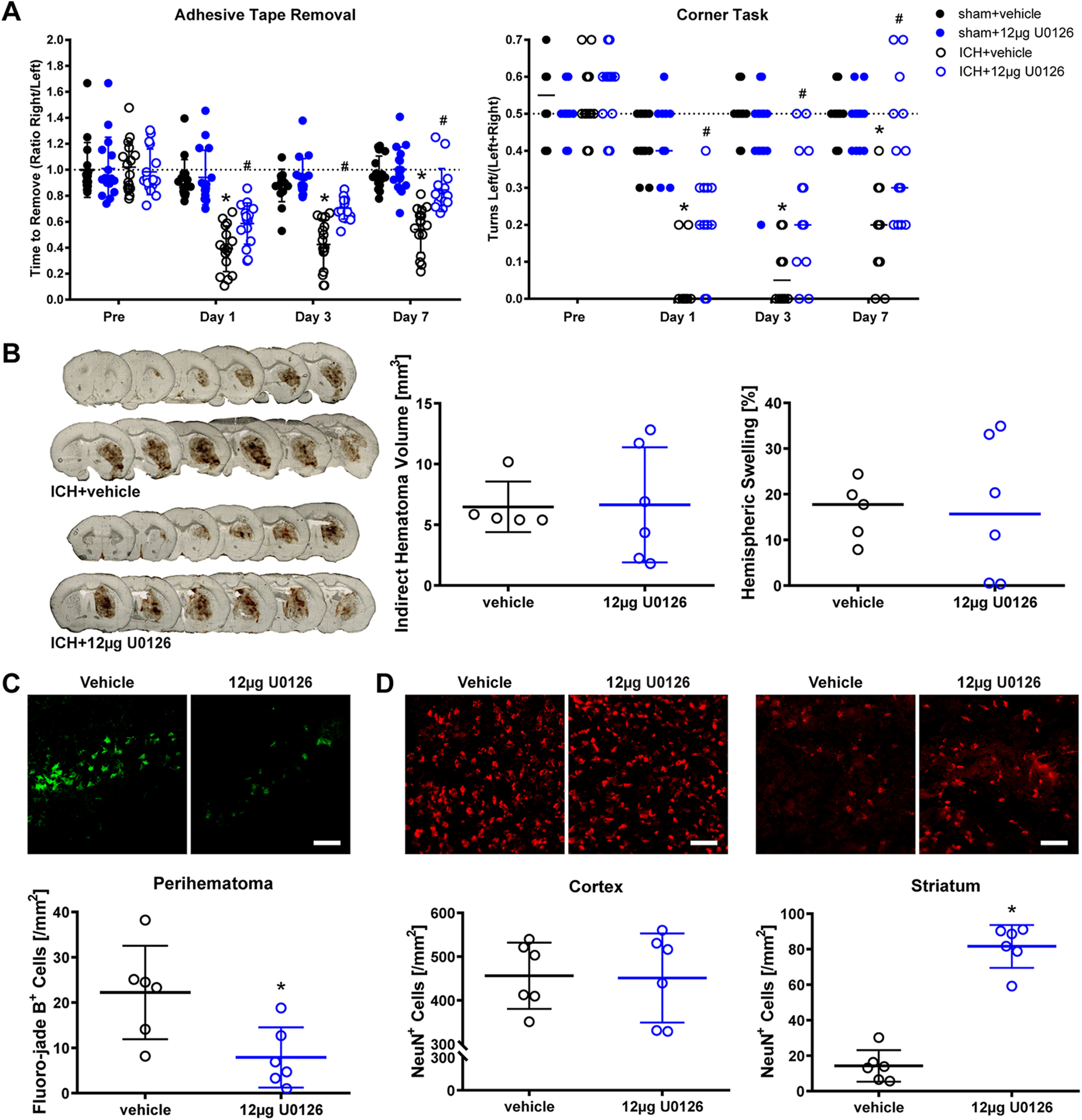
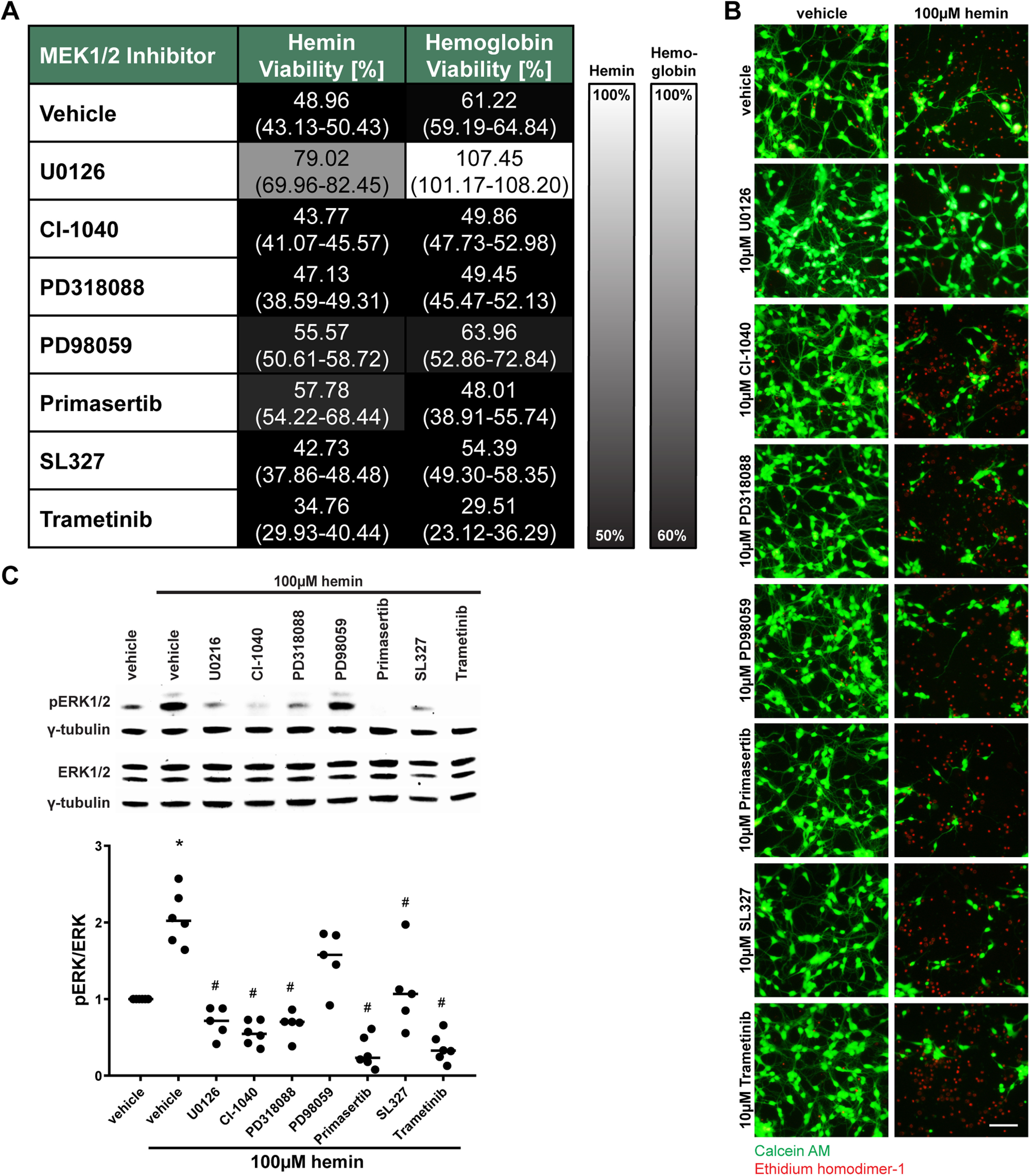
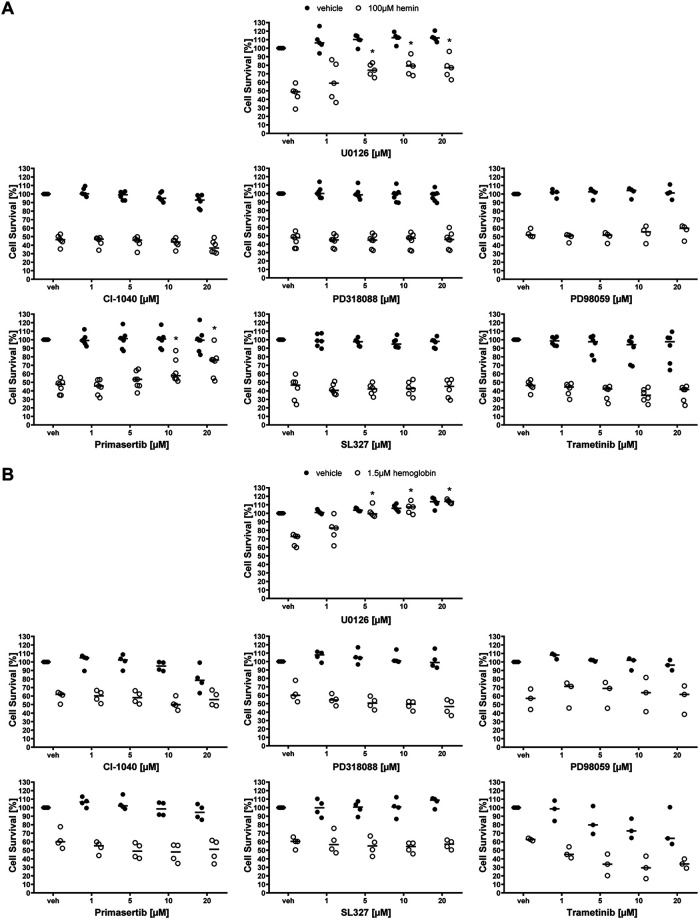
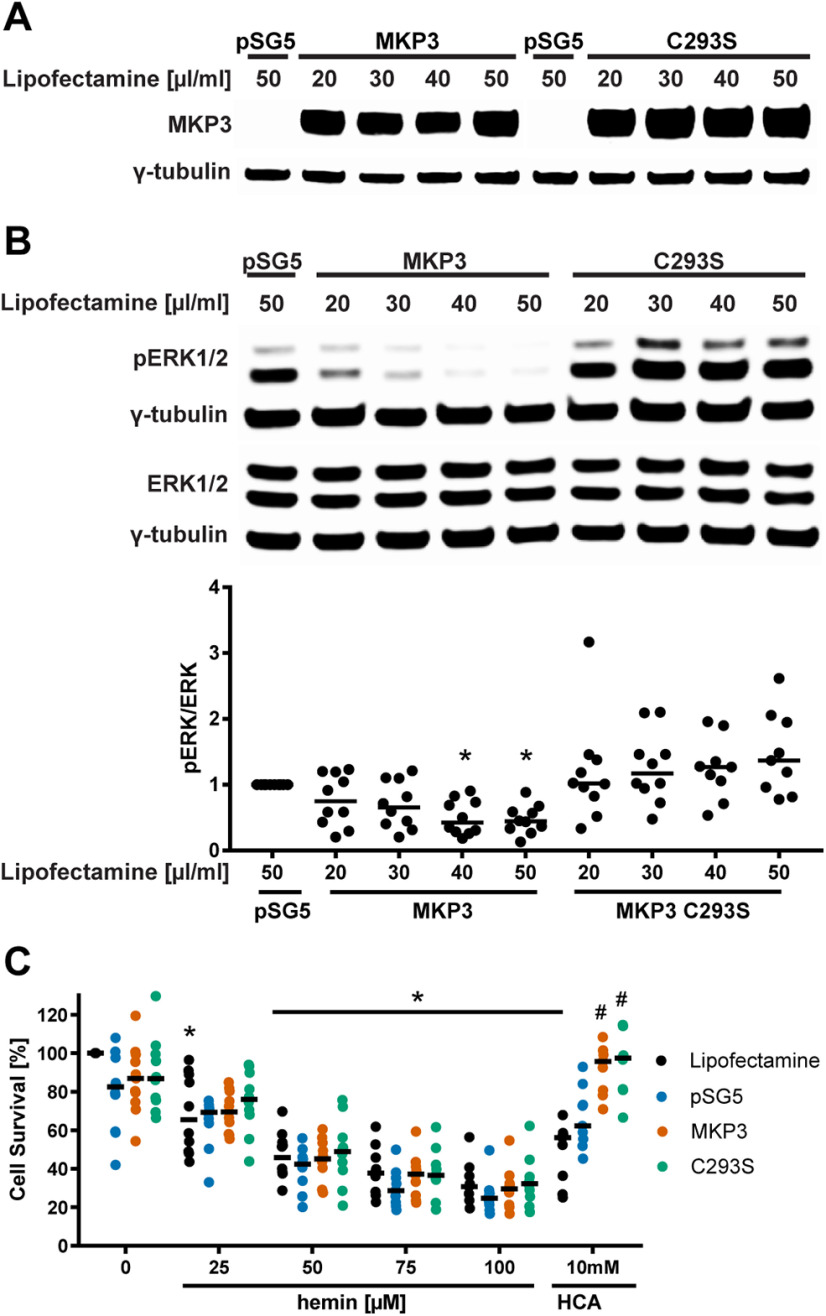
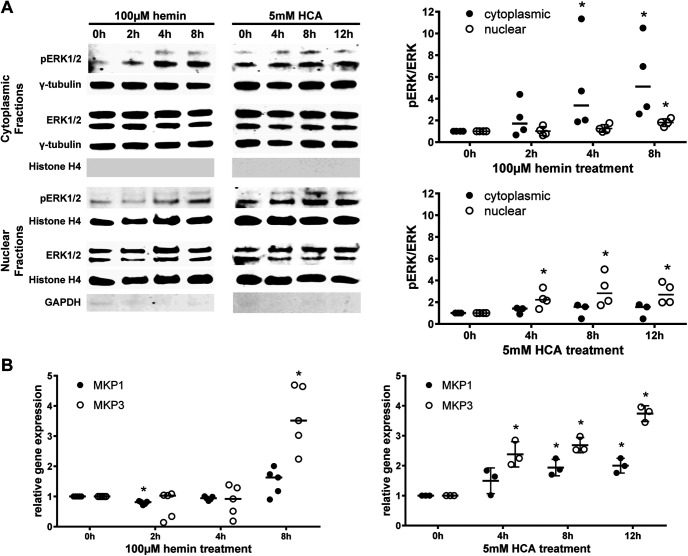
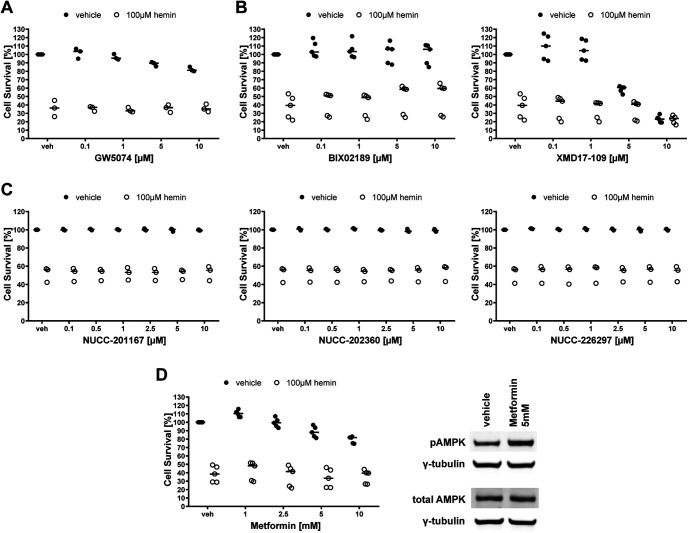
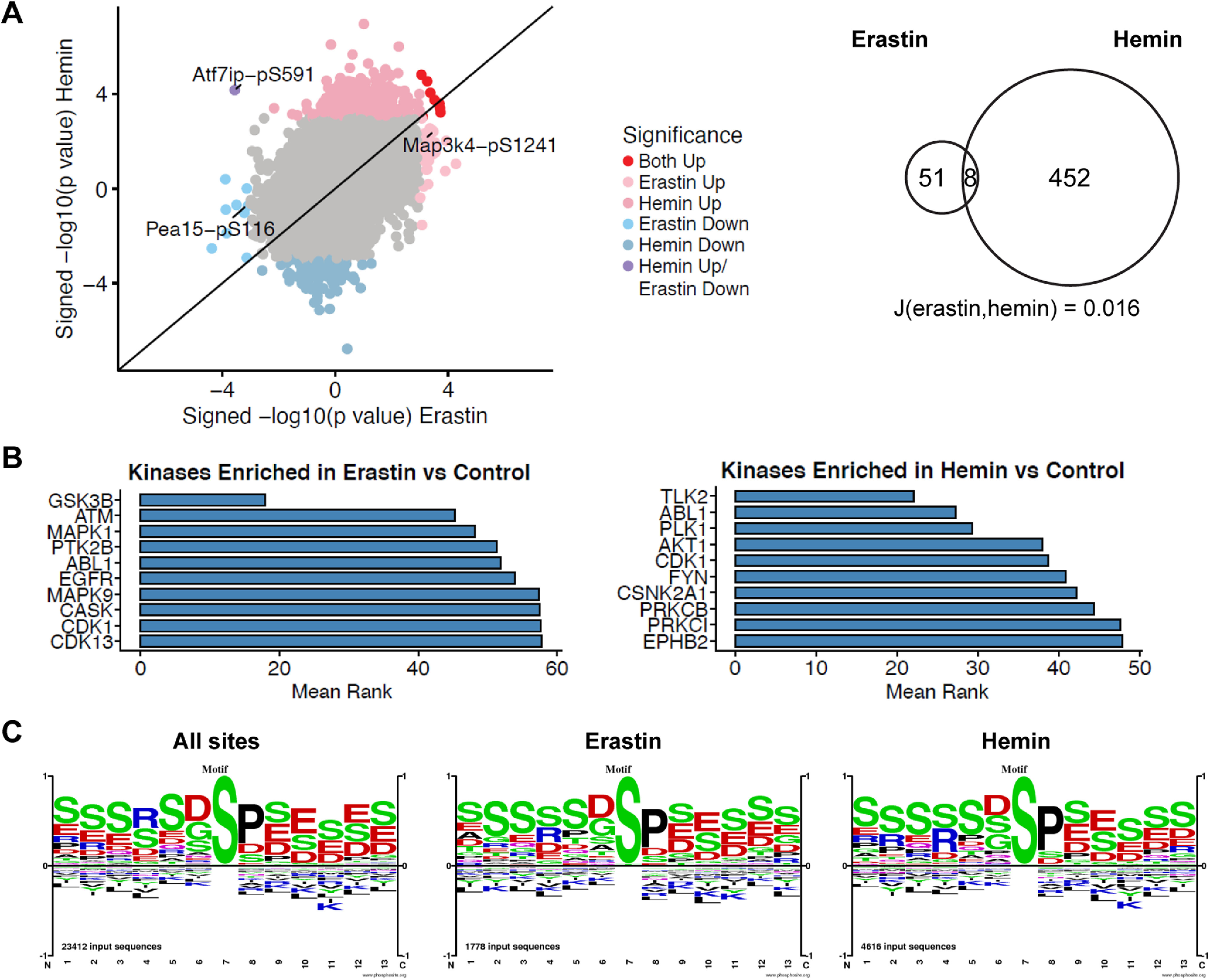
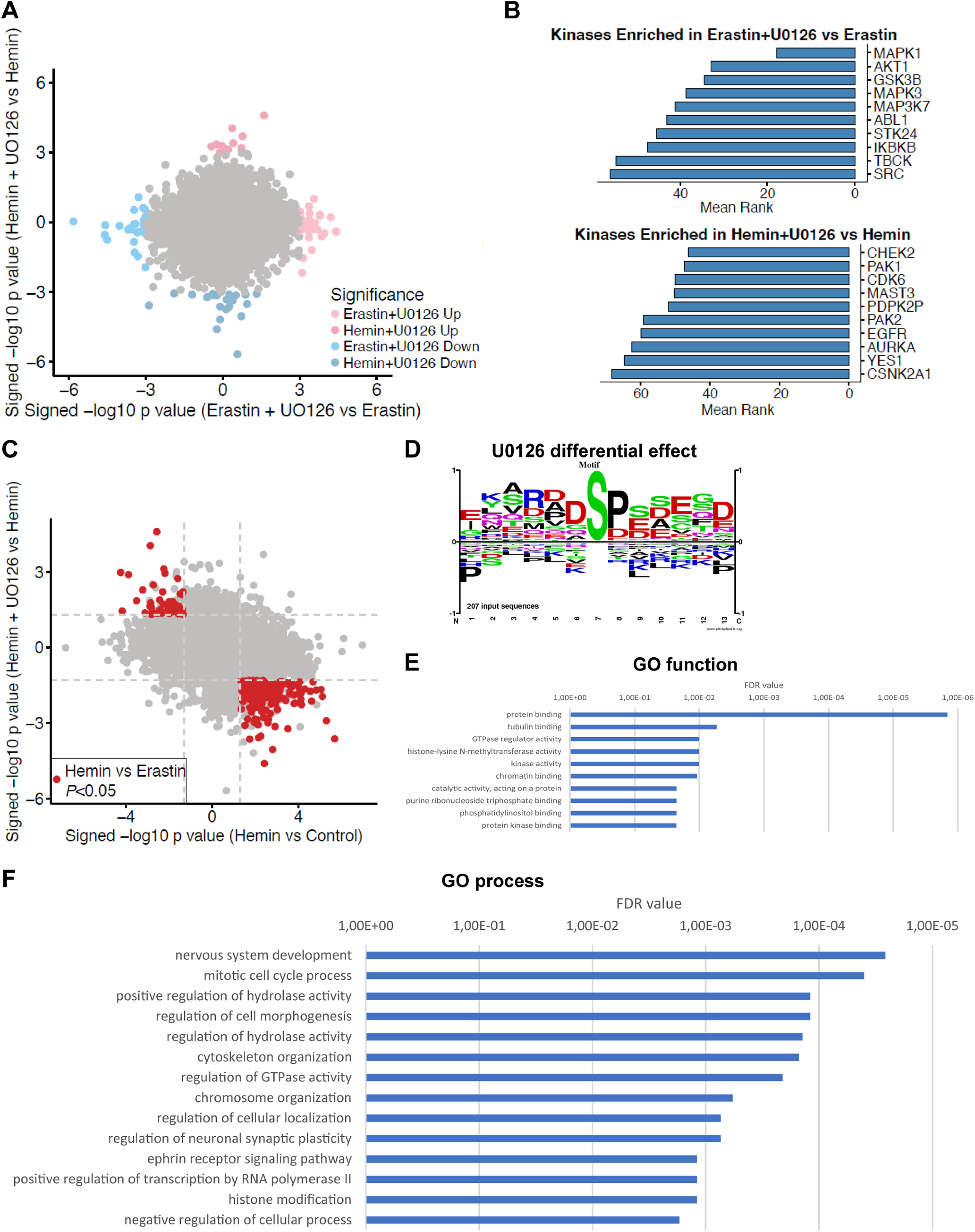
Ferroptosis is a caspase-independent, iron-dependent form of regulated necrosis extant in traumatic brain injury, Huntington disease, and hemorrhagic stroke. It can be activated by cystine deprivation leading to glutathione depletion, the insufficiency of the antioxidant glutathione peroxidase-4, and the hemolysis products hemoglobin and hemin. A cardinal feature of ferroptosis is extracellular signal-regulated kinase (ERK)1/2 activation culminating in its translocation to the nucleus. We have previously confirmed that the mitogen-activated protein (MAP) kinase kinase (MEK) inhibitor U0126 inhibits persistent ERK1/2 phosphorylation and ferroptosis. Here, we show that hemin exposure, a model of secondary injury in brain hemorrhage and ferroptosis, activated ERK1/2 in mouse neurons. Accordingly, MEK inhibitor U0126 protected against hemin-induced ferroptosis. Unexpectedly, U0126 prevented hemin-induced ferroptosis independent of its ability to inhibit ERK1/2 signaling. In contrast to classical ferroptosis in neurons or cancer cells, chemically diverse inhibitors of MEK did not block hemin-induced ferroptosis, nor did the forced expression of the ERK-selective MAP kinase phosphatase (MKP)3. We conclude that hemin or hemoglobin-induced ferroptosis, unlike glutathione depletion, is ERK1/2-independent. Together with recent studies, our findings suggest the existence of a novel subtype of neuronal ferroptosis relevant to bleeding in the brain that is 5-lipoxygenase-dependent, ERK-independent, and transcription-independent. Remarkably, our unbiased phosphoproteome analysis revealed dramatic differences in phosphorylation induced by two ferroptosis subtypes. As U0126 also reduced cell death and improved functional recovery after hemorrhagic stroke in male mice, our analysis also provides a template on which to build a search for U0126's effects in a variant of neuronal ferroptosis.SIGNIFICANCE STATEMENT Ferroptosis is an iron-dependent mechanism of regulated necrosis that has been linked to hemorrhagic stroke. Common features of ferroptotic death induced by diverse stimuli are the depletion of the antioxidant glutathione, production of lipoxygenase-dependent reactive lipids, sensitivity to iron chelation, and persistent activation of extracellular signal-regulated kinase (ERK) signaling. Unlike classical ferroptosis induced in neurons or cancer cells, here we show that ferroptosis induced by hemin is ERK-independent. Paradoxically, the canonical MAP kinase kinase (MEK) inhibitor U0126 blocks brain hemorrhage-induced death. Altogether, these data suggest that a variant of ferroptosis is unleashed in hemorrhagic stroke. We present the first, unbiased phosphoproteomic analysis of ferroptosis as a template on which to understand distinct paths to cell death that meet the definition of ferroptosis.
Keywords: MAP signaling; brain hemorrhage; cell death; ferroptosis; phosphoproteomics; stroke.
Copyright © 2022 the authors.
Figures
References
-
- Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, Hondal RJ, Mukherjee S, Cave JW, Sagdullaev BT, Karuppagounder SS, Ratan RR (2019) Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177:1262–1279.e25. 10.1016/j.cell.2019.03.032 - DOI - PubMed
-
- Basso M, Berlin J, Xia L, Sleiman SF, Ko B, Haskew-Layton R, Kim E, Antonyak MA, Cerione RA, Iismaa SE, Willis D, Cho S, Ratan RR (2012) Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci 32:6561–6569. 10.1523/JNEUROSCI.3353-11.2012 - DOI - PMC - PubMed
-
- Canals S, Casarejos MJ, de Bernardo S, Solano RM, Mena MA (2003) Selective and persistent activation of extracellular signal-regulated protein kinase by nitric oxide in glial cells induces neuronal degeneration in glutathione-depleted midbrain cultures. Mol Cell Neurosci 24:1012–1026. 10.1016/j.mcn.2003.08.004 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous