

Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients
- PMID: 34991663
- PMCID: PMC8739729
- DOI: 10.1186/s13024-021-00511-x
Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients
Abstract
Background: In response to injury, neurons activate a program of organized axon self-destruction initiated by the NAD+ hydrolase, SARM1. In healthy neurons SARM1 is autoinhibited, but single amino acid changes can abolish autoinhibition leading to constitutively active SARM1 enzymes that promote degeneration when expressed in cultured neurons.
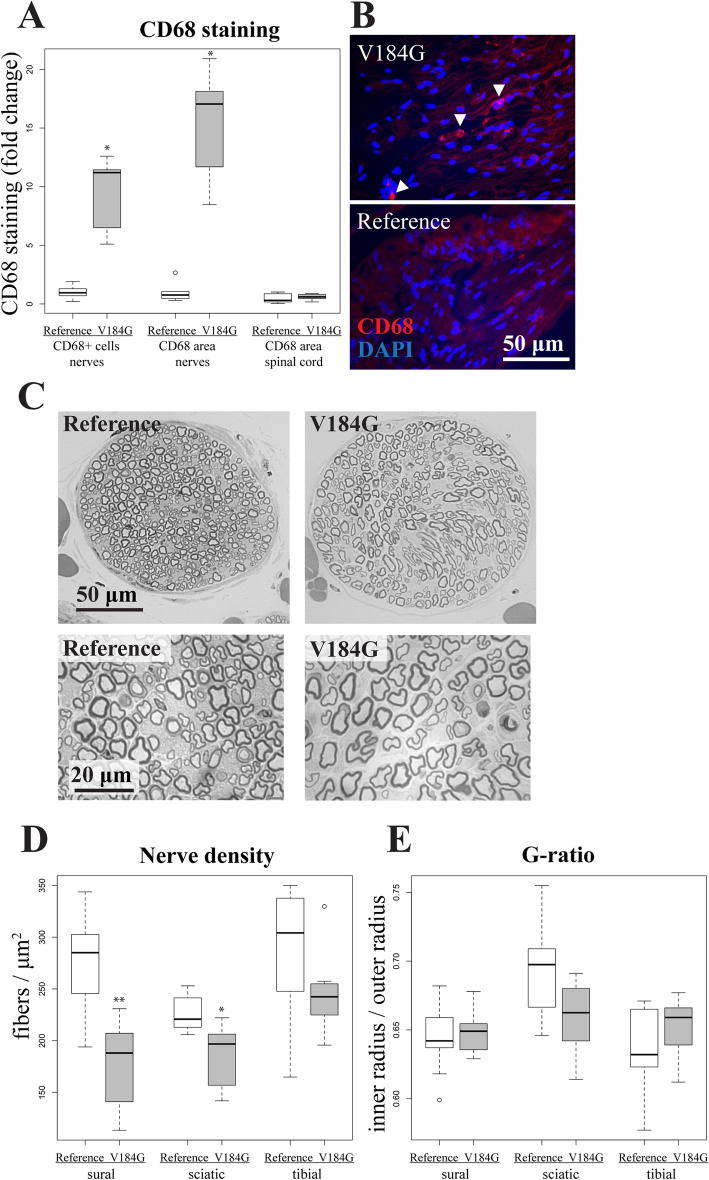
Methods: To investigate whether naturally occurring human variants might disrupt SARM1 autoinhibition and potentially contribute to risk for neurodegenerative disease, we assayed the enzymatic activity of all 42 rare SARM1 alleles identified among 8507 amyotrophic lateral sclerosis (ALS) patients and 9671 controls. We then intrathecally injected mice with virus expressing SARM1 constructs to test the capacity of an ALS-associated constitutively active SARM1 variant to promote neurodegeneration in vivo.
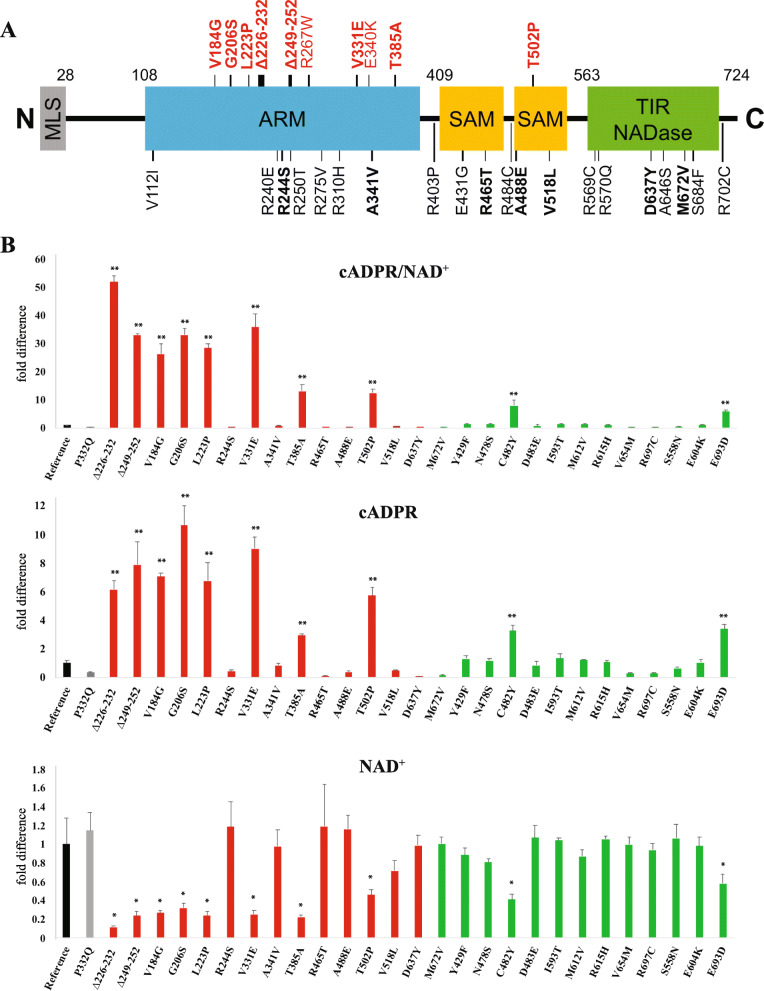
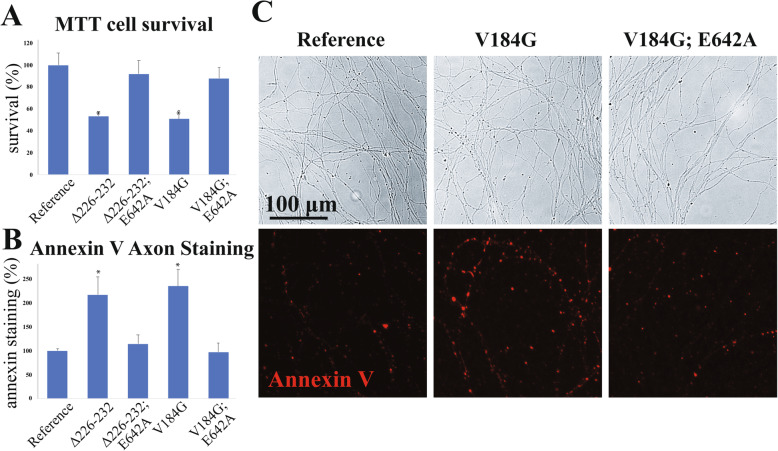
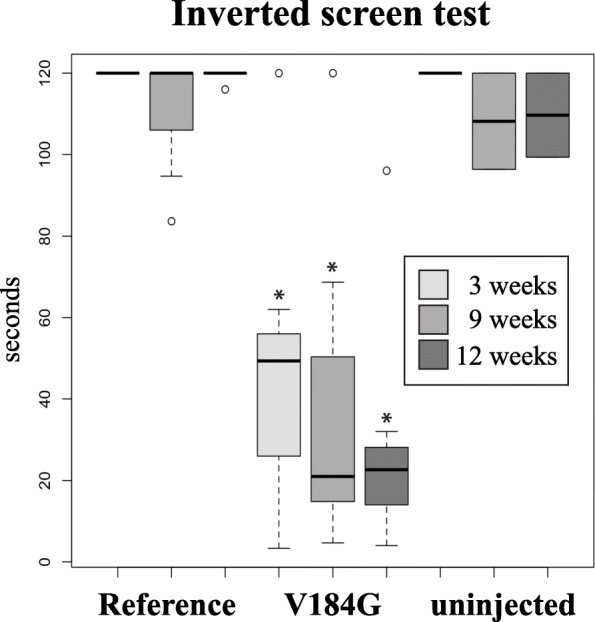
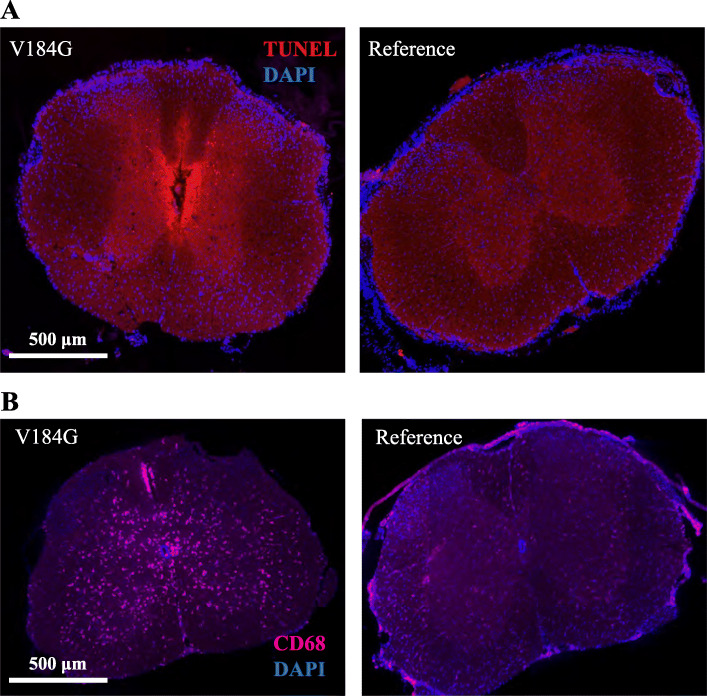
Results: Twelve out of 42 SARM1 missense variants or small in-frame deletions assayed exhibit constitutive NADase activity, including more than half of those that are unique to the ALS patients or that occur in multiple patients. There is a > 5-fold enrichment of constitutively active variants among patients compared to controls. Expression of constitutively active ALS-associated SARM1 alleles in cultured dorsal root ganglion (DRG) neurons is pro-degenerative and cytotoxic. Intrathecal injection of an AAV expressing the common SARM1 reference allele is innocuous to mice, but a construct harboring SARM1V184G, the constitutively active variant found most frequently among the ALS patients, causes axon loss, motor dysfunction, and sustained neuroinflammation.
Conclusions: These results implicate rare hypermorphic SARM1 alleles as candidate genetic risk factors for ALS and other neurodegenerative conditions.
Keywords: ALS; Axon; Human genetics; NAD; Neurodegeneration; Neuropathy; SARM1.
© 2022. The Author(s).
Conflict of interest statement
A.D. and J.M. are co-founders, scientific advisory board members and shareholders of Disarm Therapeutics, a wholly owned subsidiary of Eli Lilly. A.J.B. and Y.S. are consultants to Disarm Therapeutics. The authors have no other competing conflicts or financial interests.
Figures
References
-
- Lattante S, Doronzio PN, Marangi G, Conte A, Bisogni G, Bernardo D, Russo T, Lamberti D, Patrizi S, Apollo FP, Lunetta C, Scarlino S, Pozzi L, Zollino M, Riva N, Sabatelli M. Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol Aging. 2019;84:239.e9–239.e14. doi: 10.1016/j.neurobiolaging.2019.03.010. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
