

Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies
- PMID: 34992268
- PMCID: PMC9245609
- DOI: 10.1038/s41588-021-00976-y
Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies
Abstract
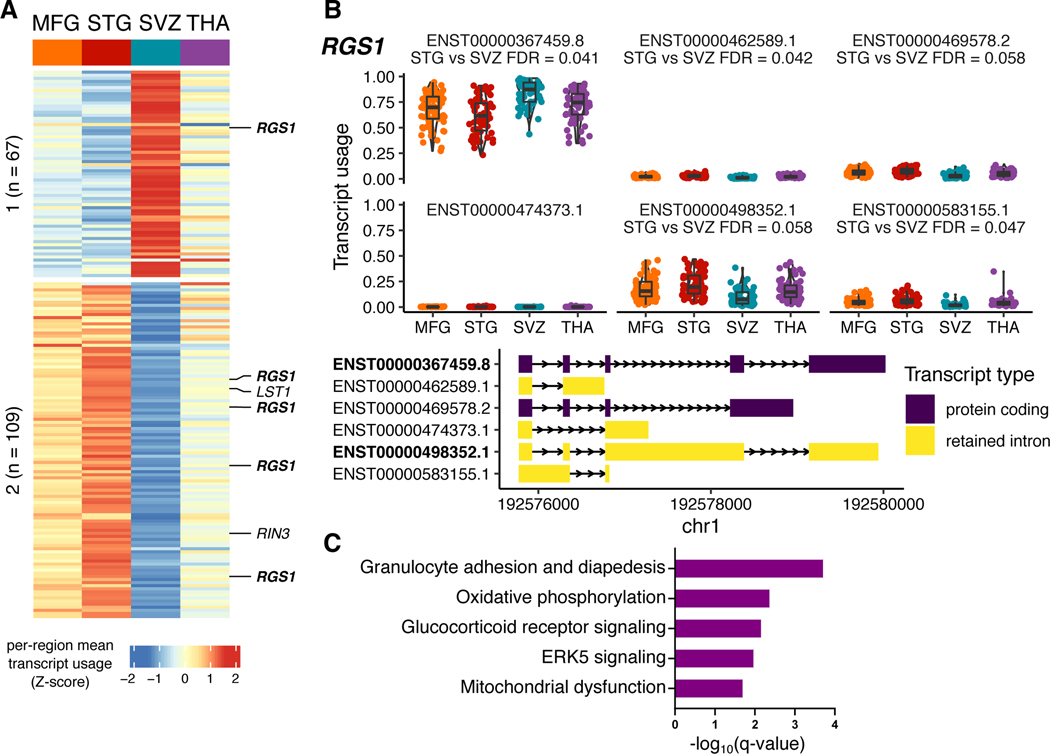
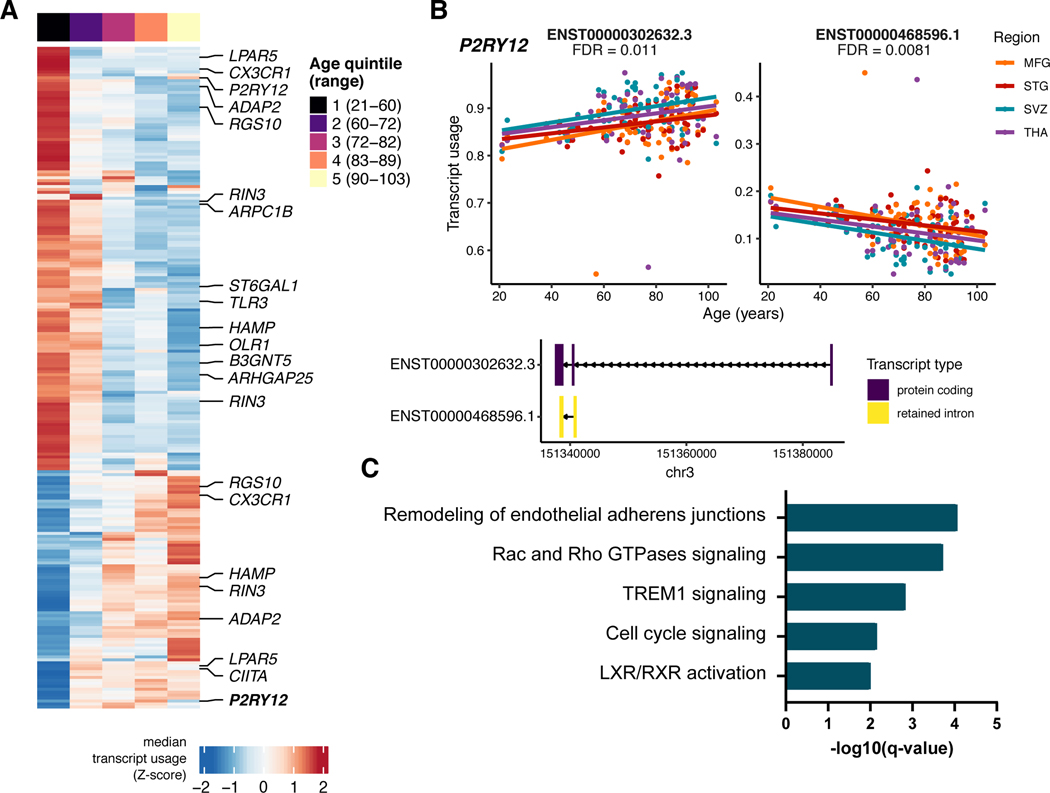
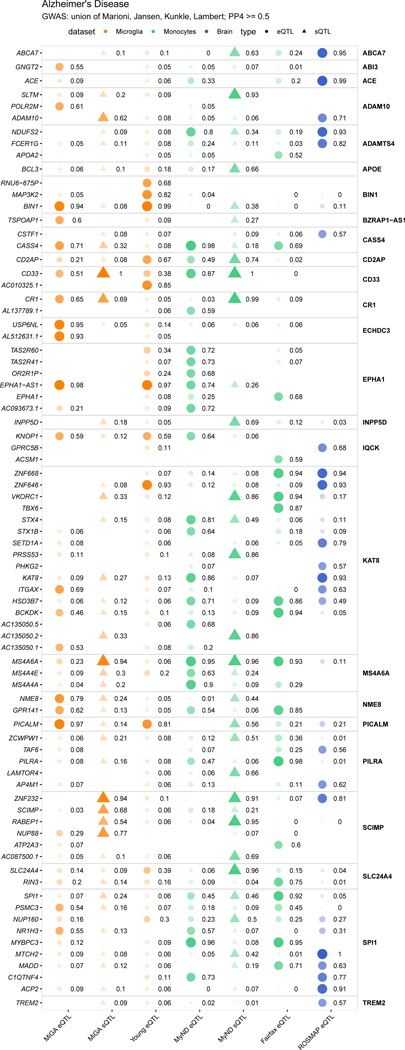
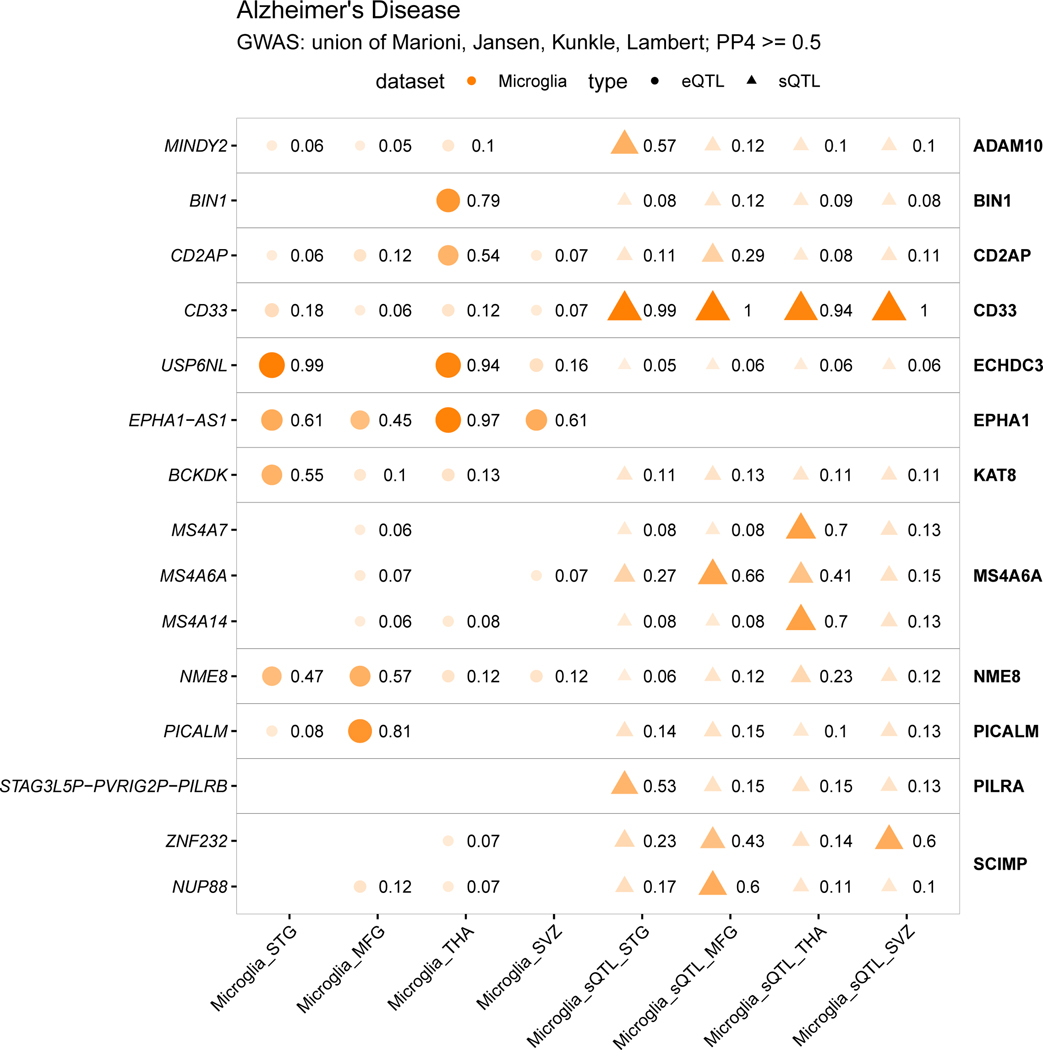
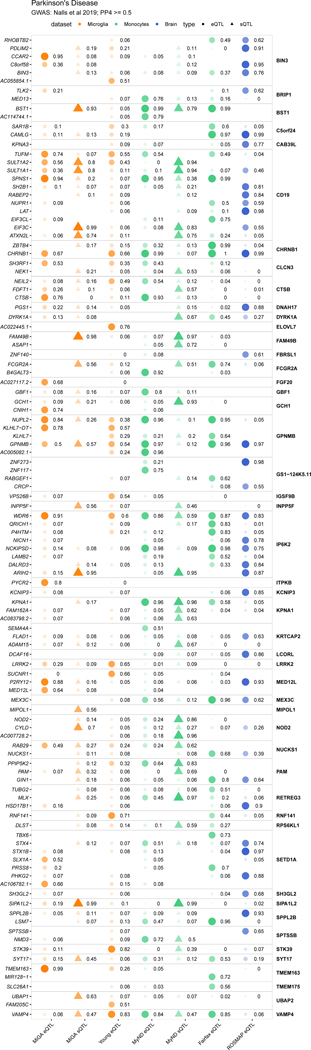
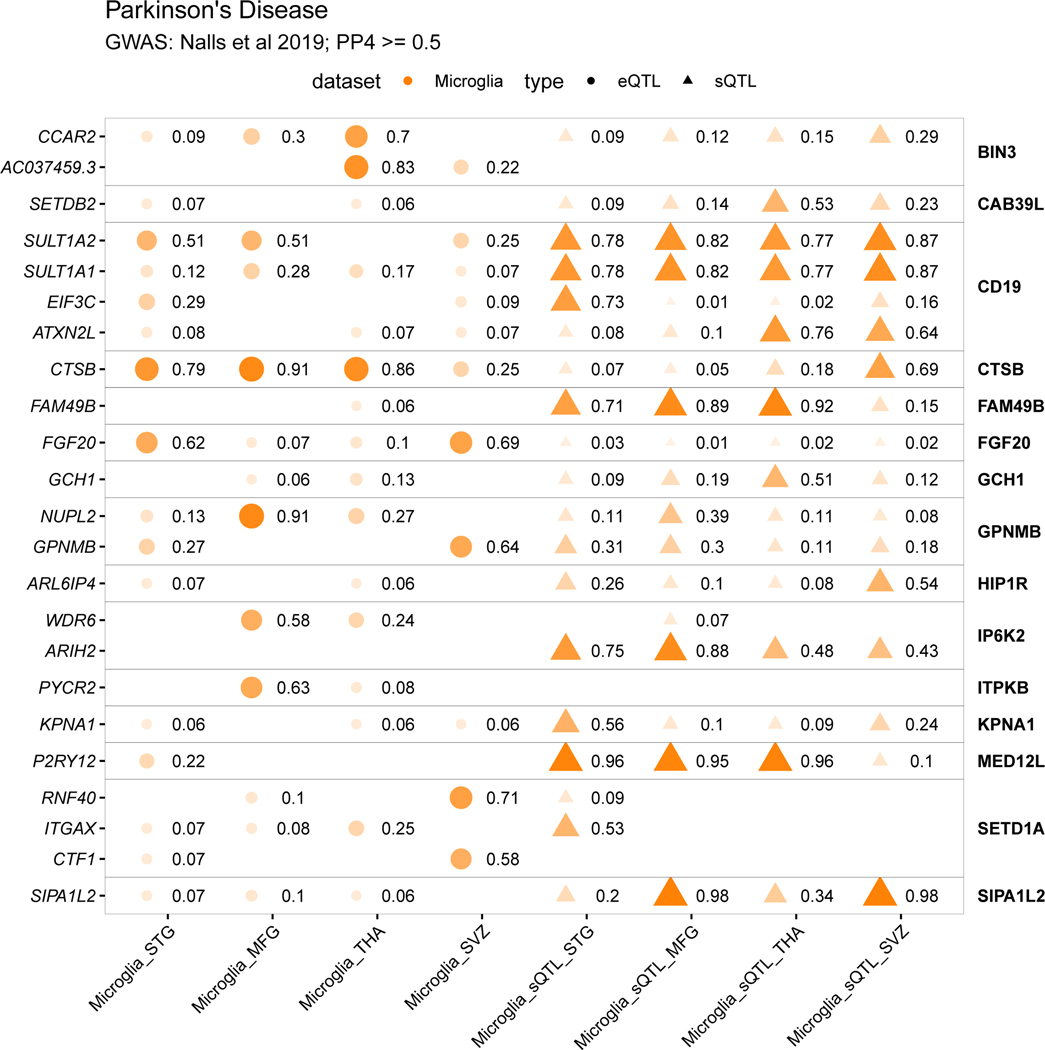
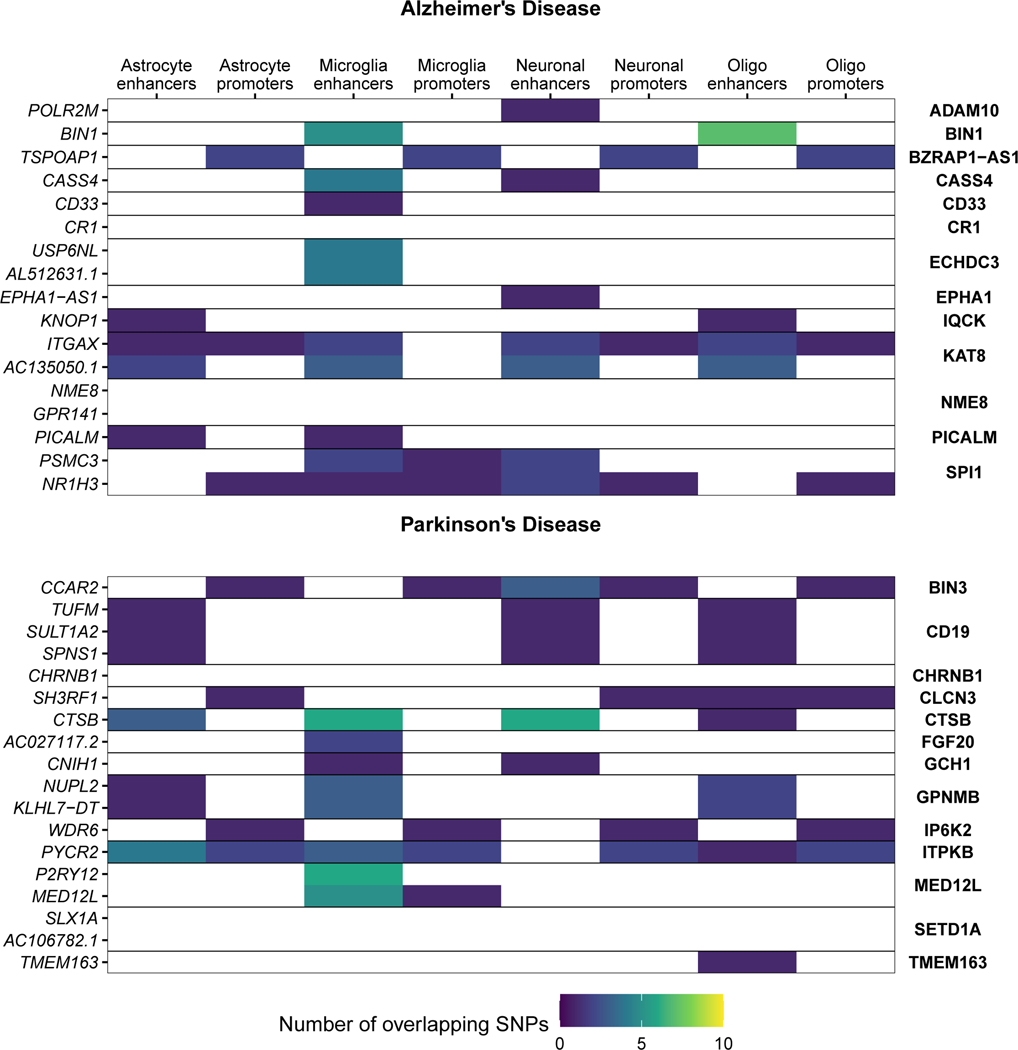
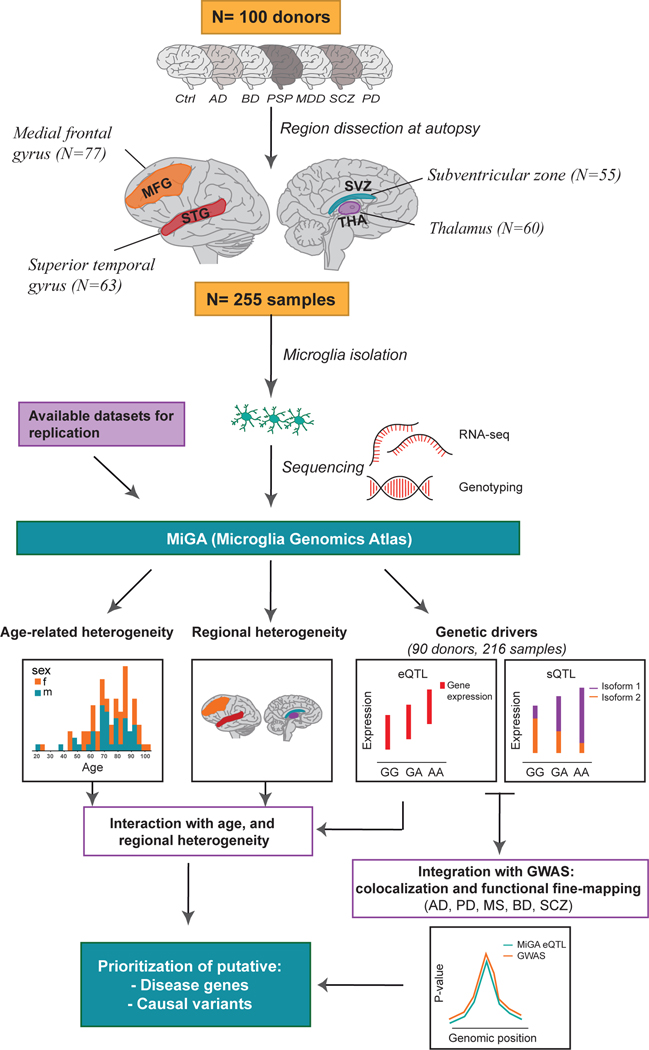
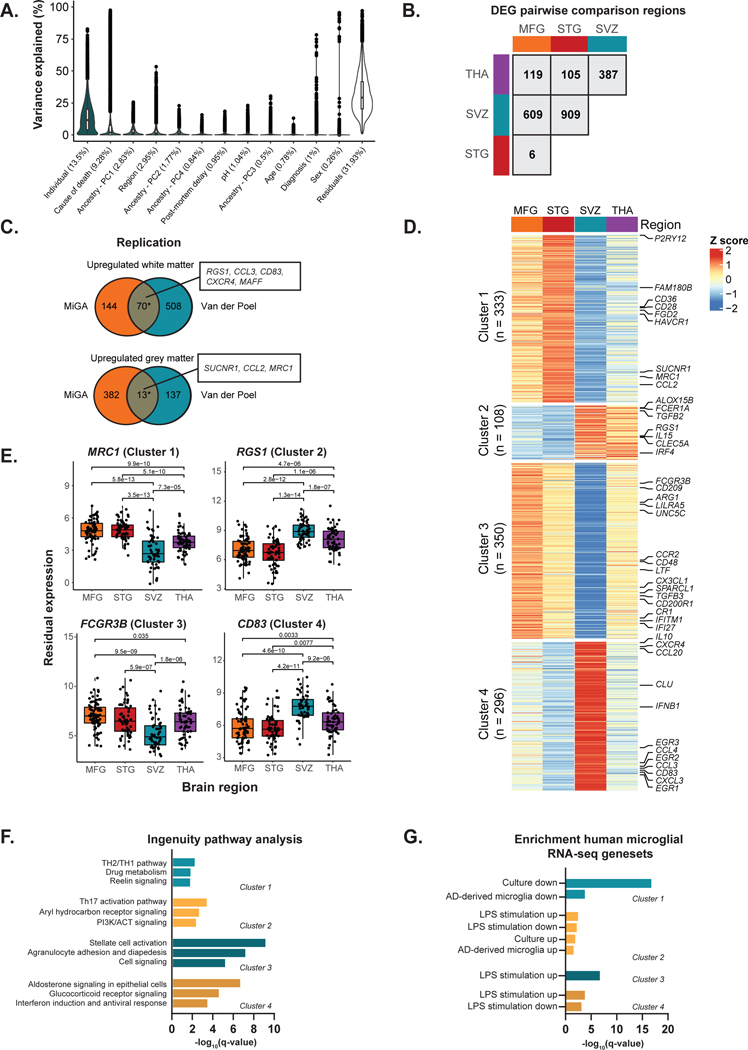
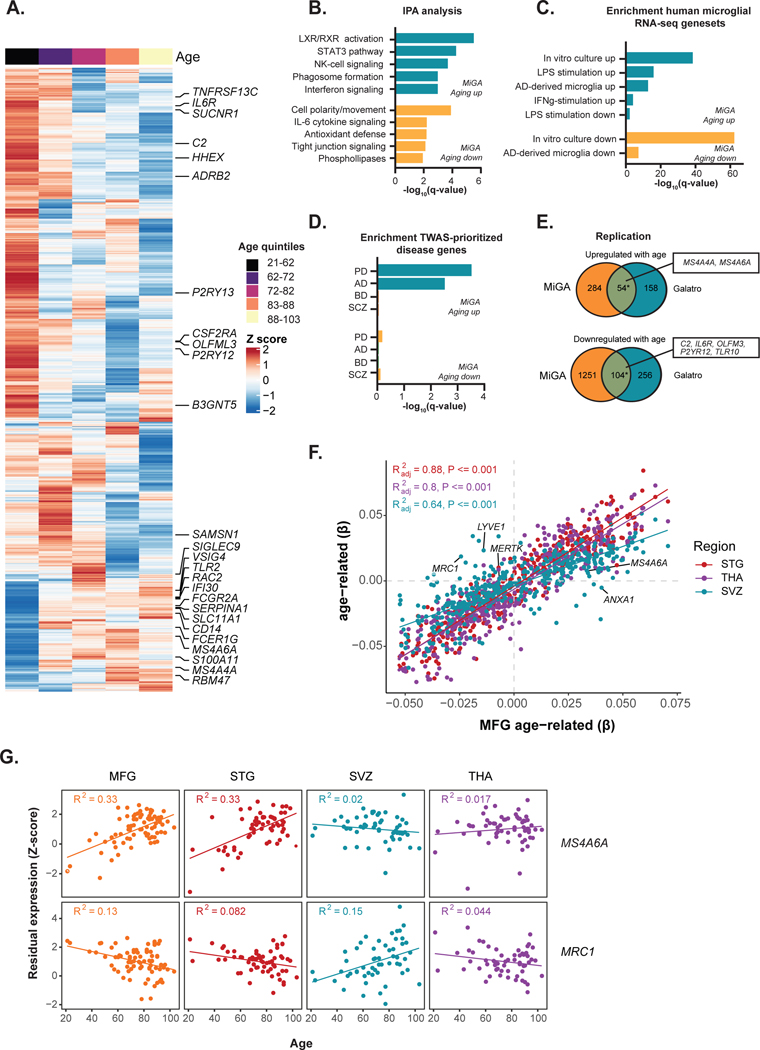
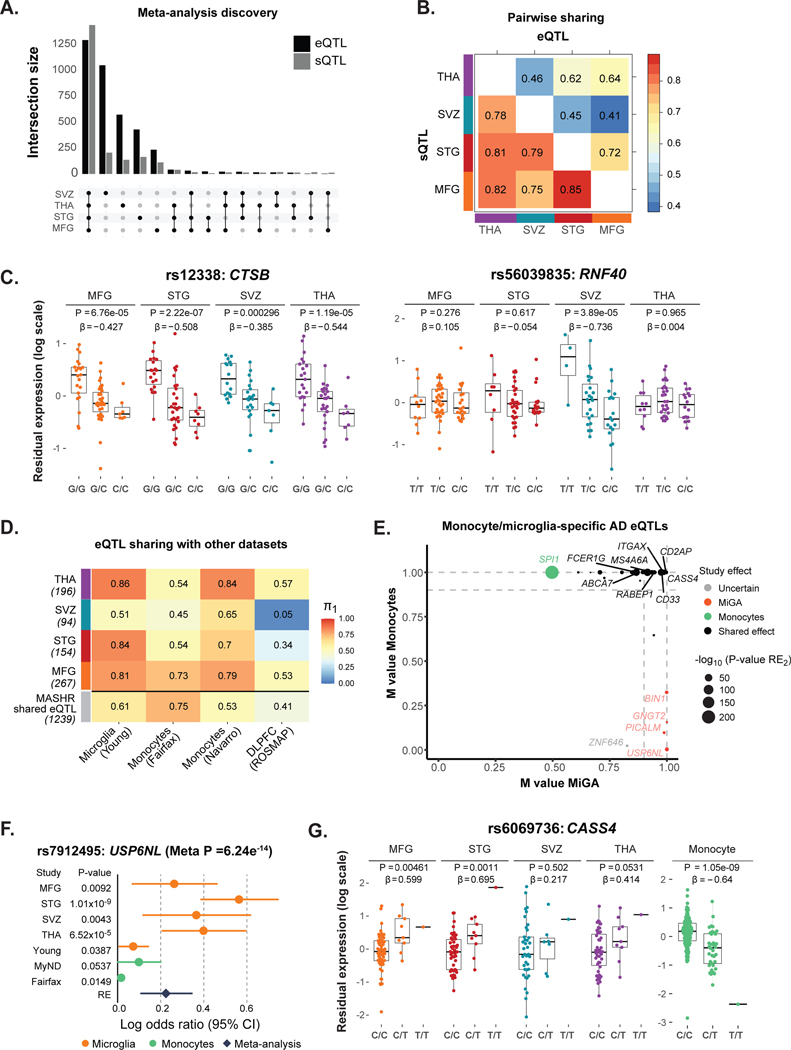
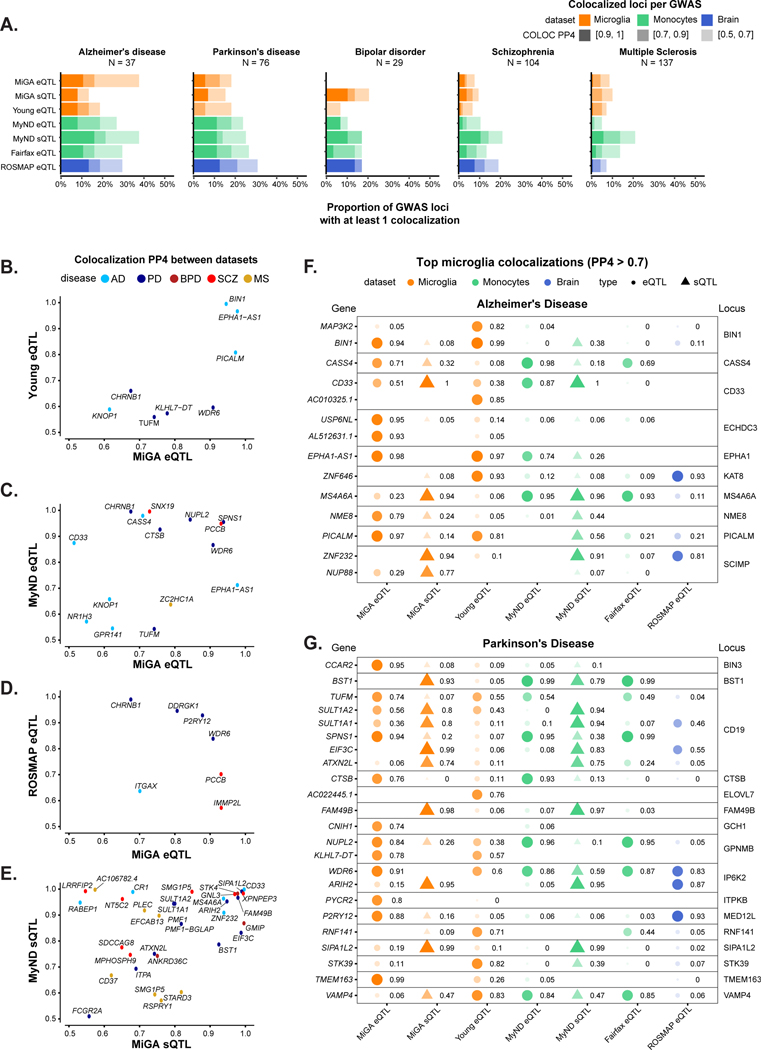
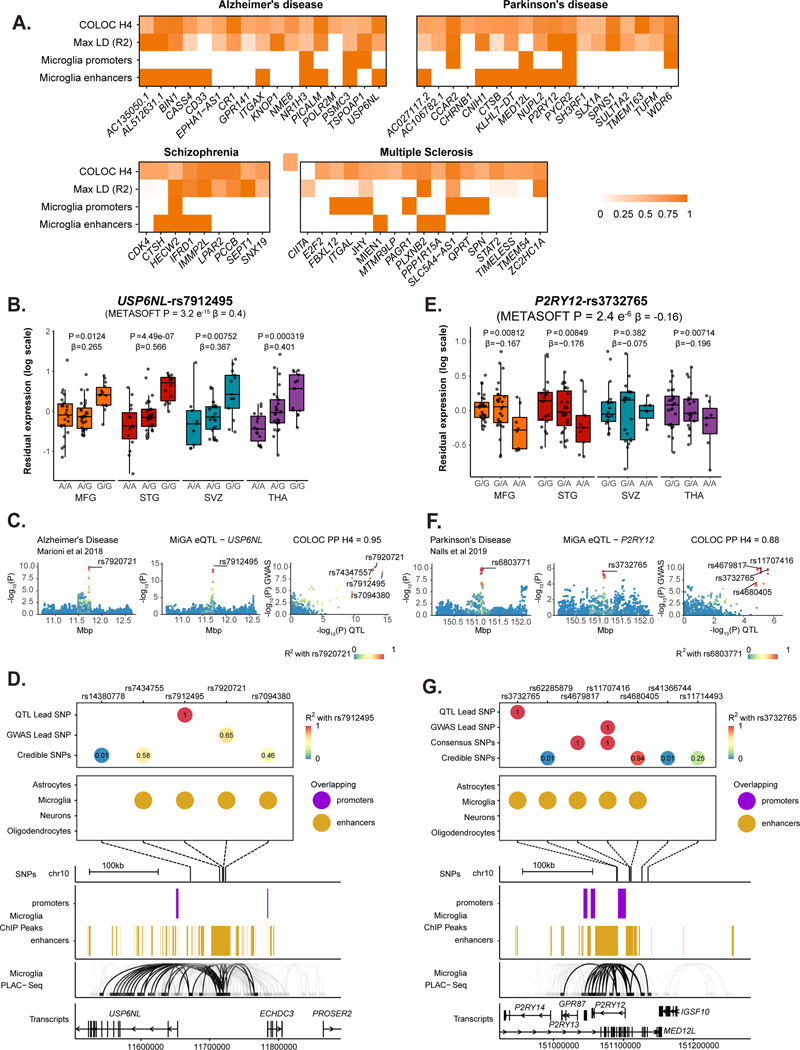
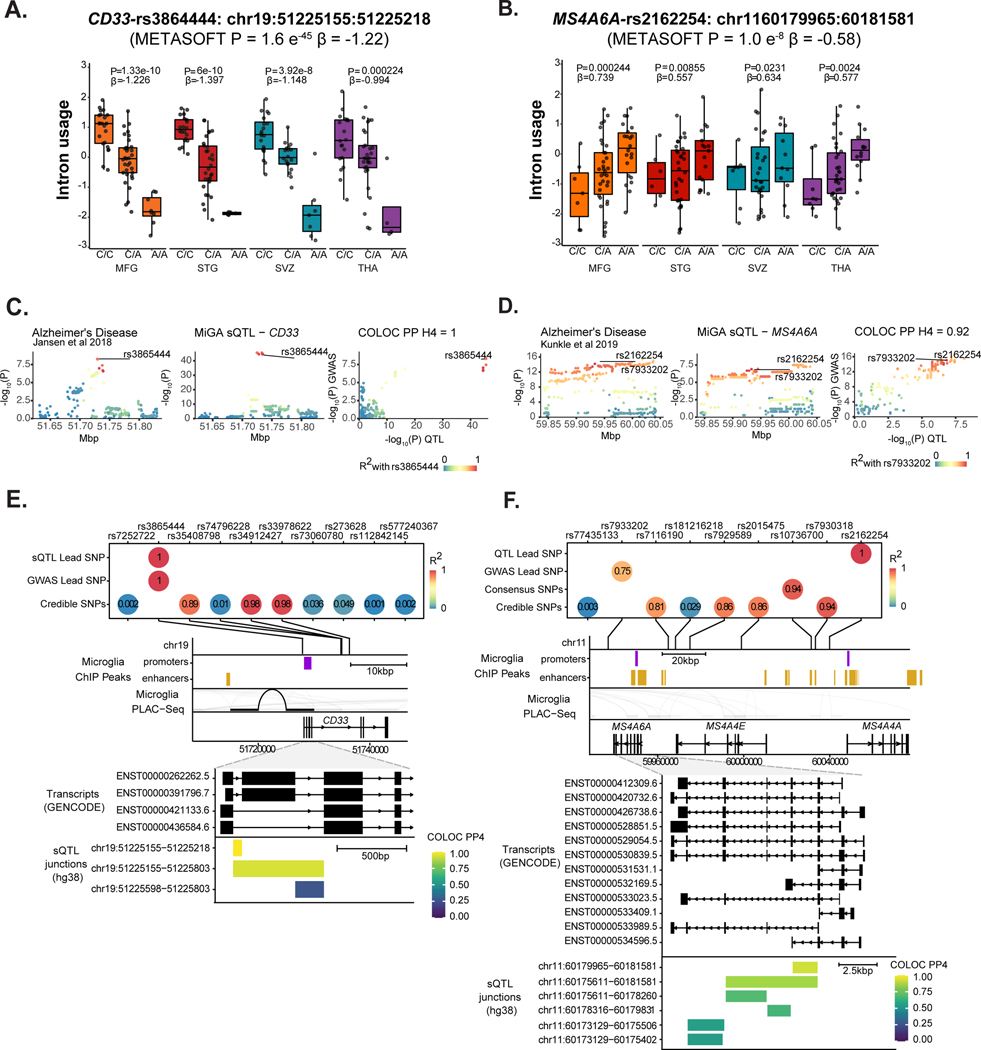
Microglia have emerged as important players in brain aging and pathology. To understand how genetic risk for neurological and psychiatric disorders is related to microglial function, large transcriptome studies are essential. Here we describe the transcriptome analysis of 255 primary human microglial samples isolated at autopsy from multiple brain regions of 100 individuals. We performed systematic analyses to investigate various aspects of microglial heterogeneities, including brain region and aging. We mapped expression and splicing quantitative trait loci and showed that many neurological disease susceptibility loci are mediated through gene expression or splicing in microglia. Fine-mapping of these loci nominated candidate causal variants that are within microglia-specific enhancers, finding associations with microglial expression of USP6NL for Alzheimer's disease and P2RY12 for Parkinson's disease. We have built the most comprehensive catalog to date of genetic effects on the microglial transcriptome and propose candidate functional variants in neurological and psychiatric disorders.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
Comment in
-
Microglial transcriptomics meets genetics: new disease leads.Nat Rev Neurol. 2022 Apr;18(4):191-192. doi: 10.1038/s41582-022-00633-w. Nat Rev Neurol. 2022. PMID: 35228701 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
