

IDH-mutated gliomas promote epileptogenesis through d-2-hydroxyglutarate-dependent mTOR hyperactivation
- PMID: 34994387
- PMCID: PMC9435503
- DOI: 10.1093/neuonc/noac003
IDH-mutated gliomas promote epileptogenesis through d-2-hydroxyglutarate-dependent mTOR hyperactivation
Abstract
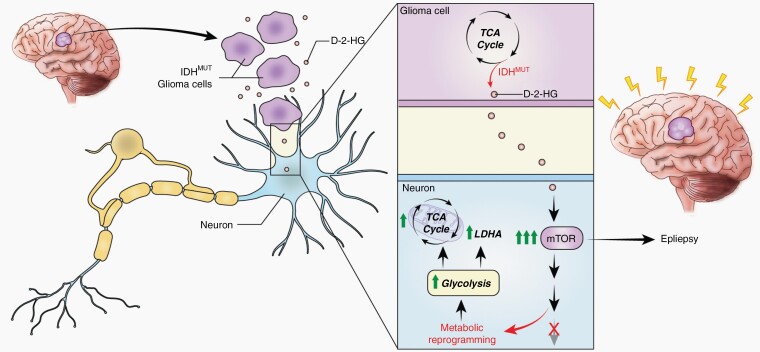
Background: Uncontrolled seizures in patients with gliomas have a significant impact on quality of life and morbidity, yet the mechanisms through which these tumors cause seizures remain unknown. Here, we hypothesize that the active metabolite d-2-hydroxyglutarate (d-2-HG) produced by the IDH-mutant enzyme leads to metabolic disruptions in surrounding cortical neurons that consequently promote seizures.
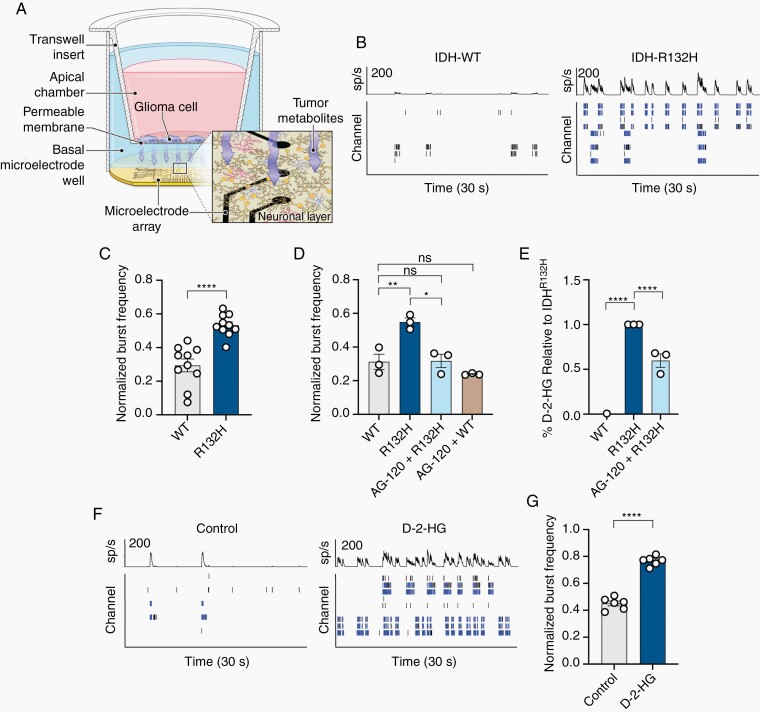
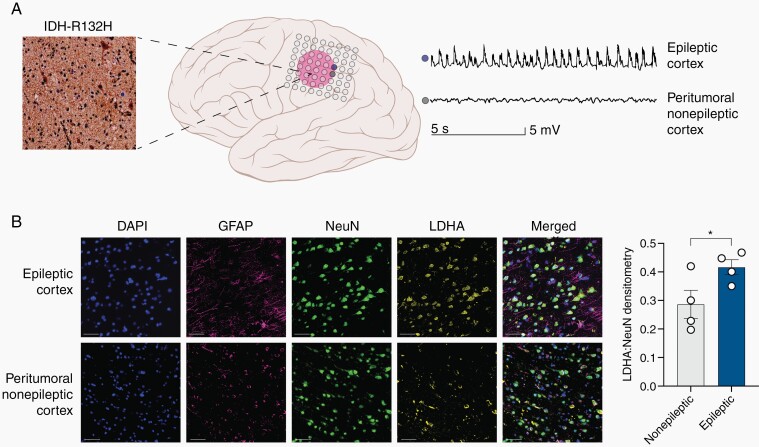
Methods: We use a complementary study of in vitro neuron-glial cultures and electrographically sorted human cortical tissue from patients with IDH-mutant gliomas to test this hypothesis. We utilize micro-electrode arrays for in vitro electrophysiological studies in combination with pharmacological manipulations and biochemical studies to better elucidate the impact of d-2-HG on cortical metabolism and neuronal spiking activity.
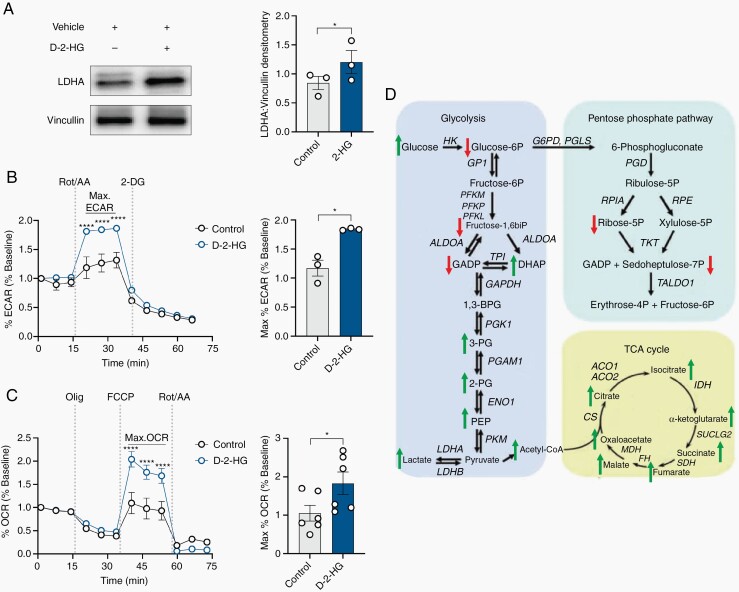
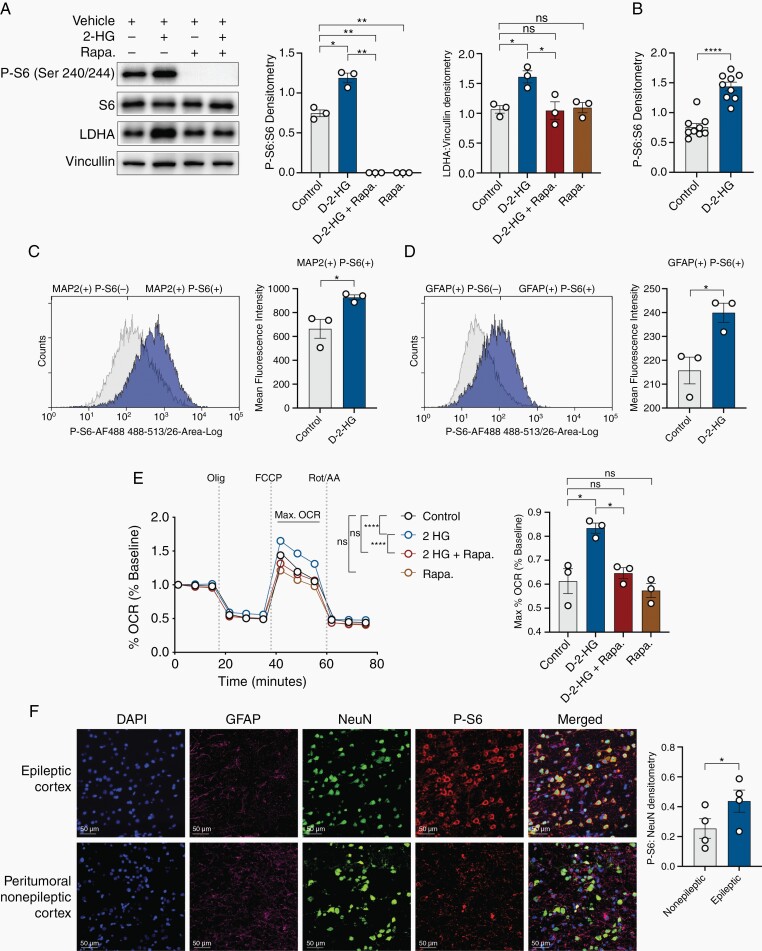
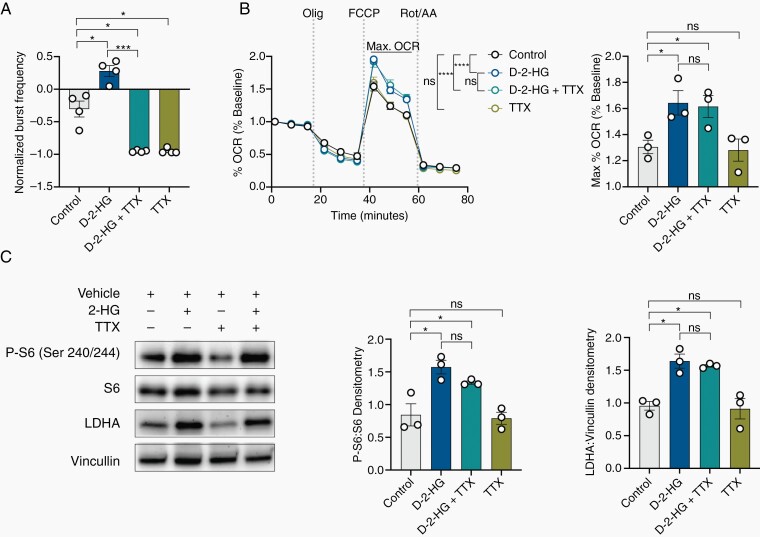
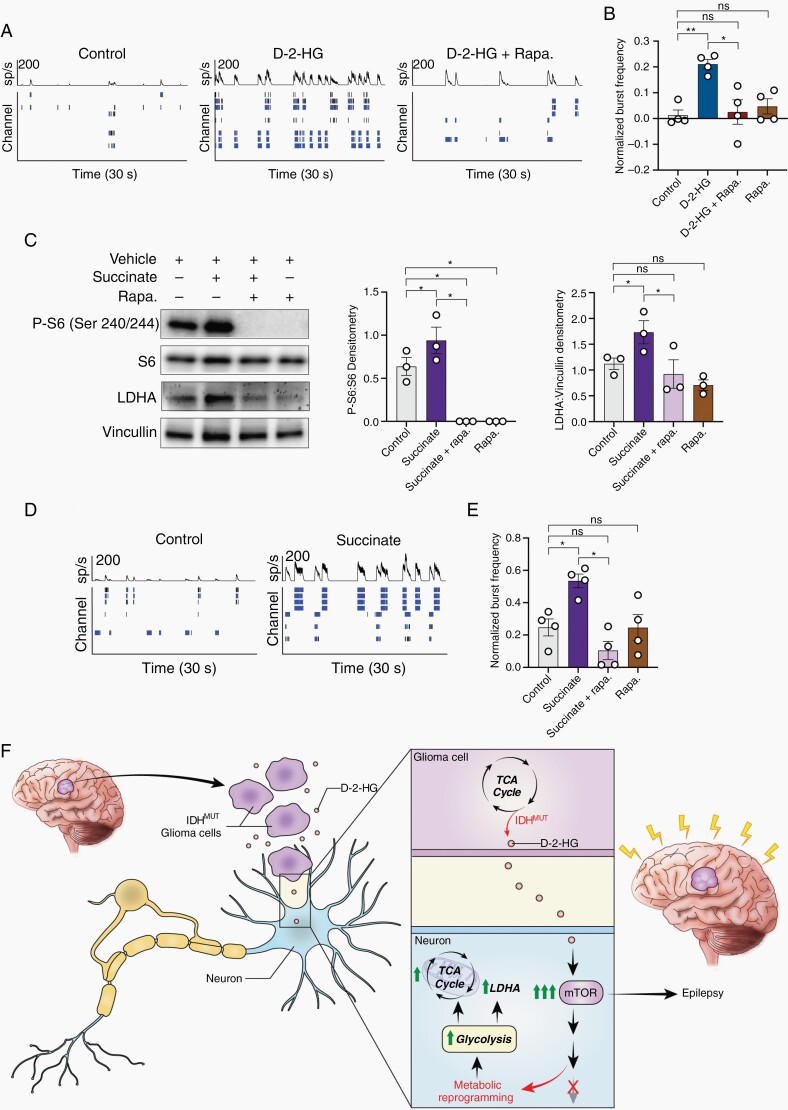
Results: We demonstrate that d-2-HG leads to increased neuronal spiking activity and promotes a distinct metabolic profile in surrounding neurons, evidenced by distinct metabolomic shifts and increased LDHA expression, as well as upregulation of mTOR signaling. The increases in neuronal activity are induced by mTOR activation and reversed with mTOR inhibition.
Conclusion: Together, our data suggest that metabolic disruptions in the surrounding cortex due to d-2-HG may be a driving event for epileptogenesis in patients with IDH-mutant gliomas.
Keywords: D-2-HG; IDH-mutated gliomas; Tumor-related epilepsy; mTOR.
Published by Oxford University Press on behalf of the Society for Neuro-Oncology 2022.
Figures
Comment in
-
Understanding epilepsy in IDH-mutated gliomas: towards a targeted therapy.Neuro Oncol. 2022 Sep 1;24(9):1436-1437. doi: 10.1093/neuonc/noac122. Neuro Oncol. 2022. PMID: 35533066 Free PMC article. No abstract available.
References
-
- Huberfeld G, Vecht CJ. Seizures and gliomas—towards a single therapeutic approach. Nat Rev Neurol. 2016;12(4):204–216. - PubMed
-
- Stockhammer F, Misch M, Helms HJ, et al. . IDH1/2 mutations in WHO grade II astrocytomas associated with localization and seizure as the initial symptom. Seizure 2012;21(3):194–197. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
