

The embryonic transcriptome of Parhyale hawaiensis reveals different dynamics of microRNAs and mRNAs during the maternal-zygotic transition
- PMID: 34996916
- PMCID: PMC8741983
- DOI: 10.1038/s41598-021-03642-9
The embryonic transcriptome of Parhyale hawaiensis reveals different dynamics of microRNAs and mRNAs during the maternal-zygotic transition
Abstract
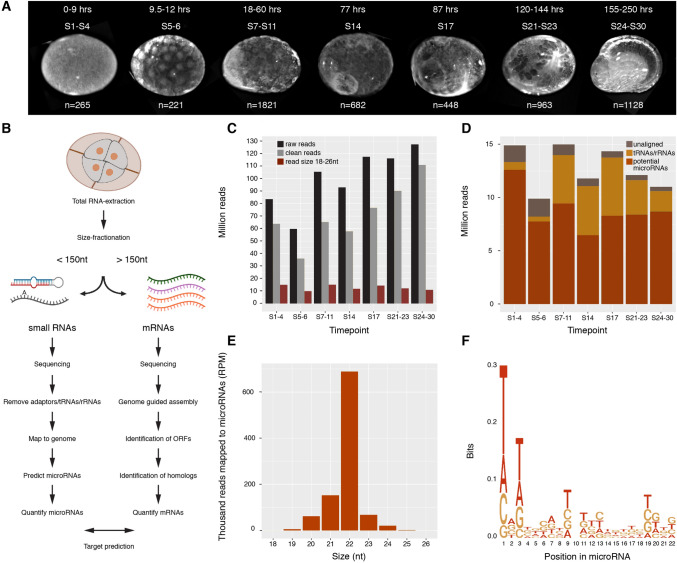
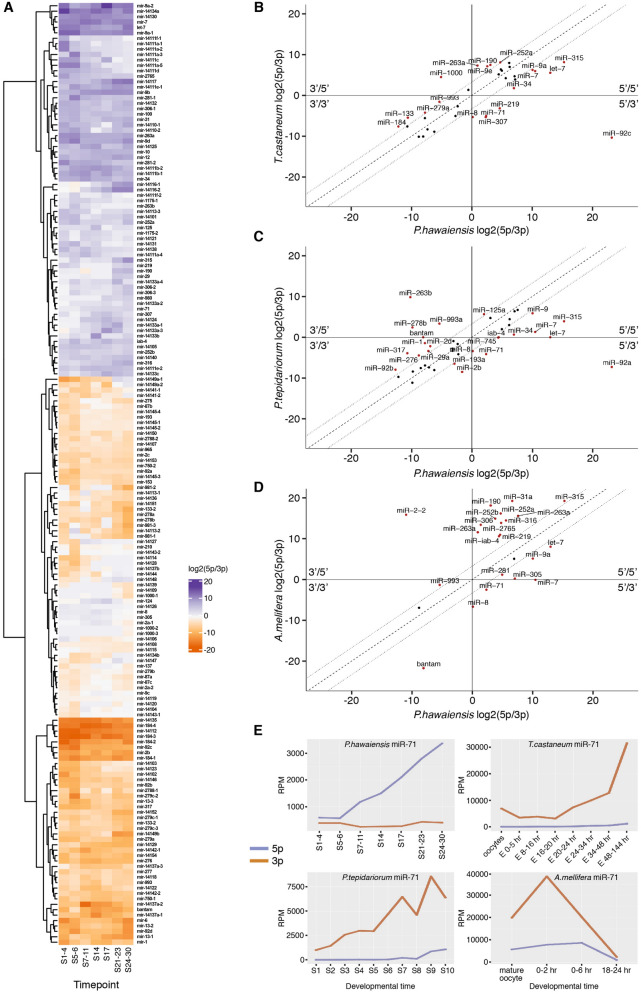
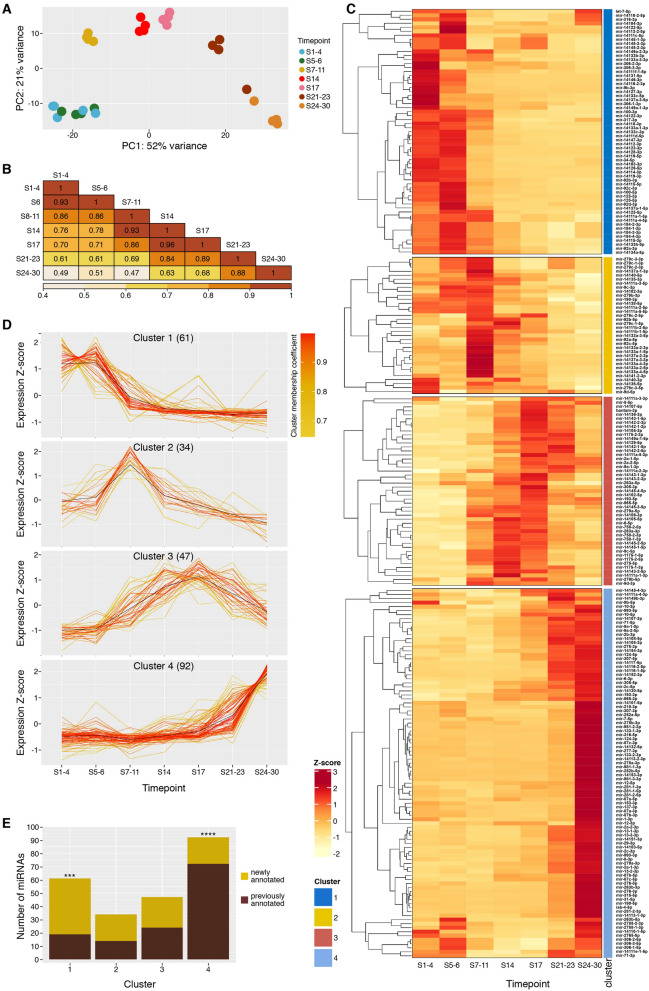
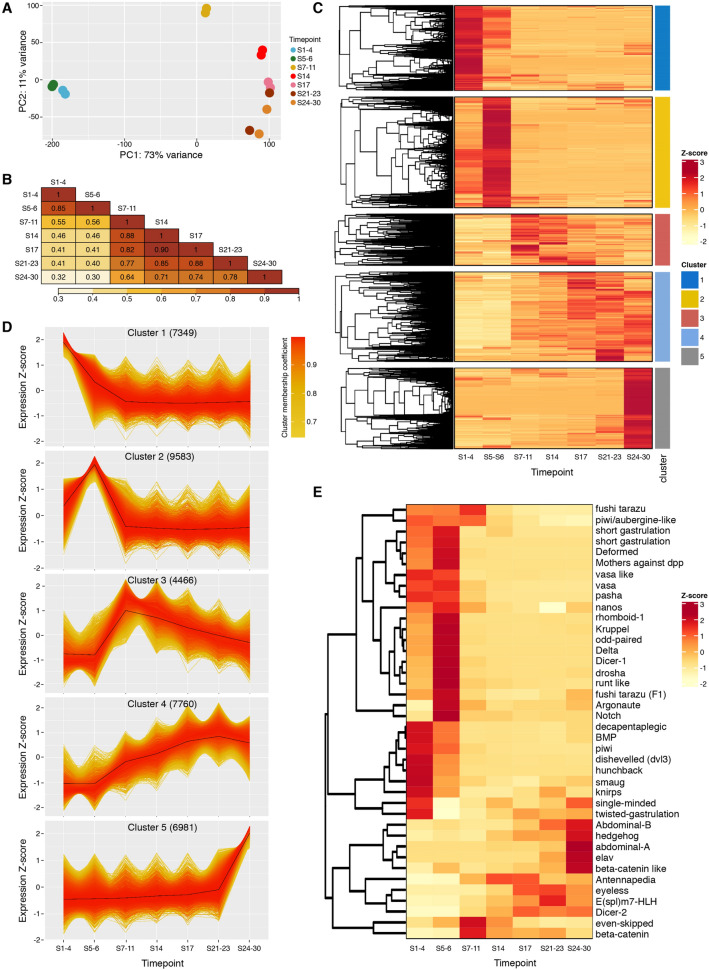
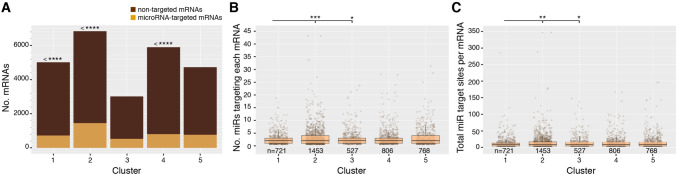
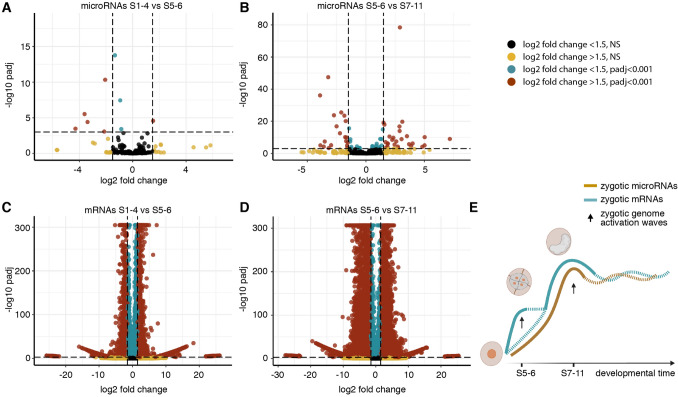
Parhyale hawaiensis has emerged as the crustacean model of choice due to its tractability, ease of imaging, sequenced genome, and development of CRISPR/Cas9 genome editing tools. However, transcriptomic datasets spanning embryonic development are lacking, and there is almost no annotation of non-protein-coding RNAs, including microRNAs. We have sequenced microRNAs, together with mRNAs and long non-coding RNAs, in Parhyale using paired size-selected RNA-seq libraries at seven time-points covering important transitions in embryonic development. Focussing on microRNAs, we annotate 175 loci in Parhyale, 88 of which have no known homologs. We use these data to annotate the microRNAome of 37 crustacean genomes, and suggest a core crustacean microRNA set of around 61 sequence families. We examine the dynamic expression of microRNAs and mRNAs during the maternal-zygotic transition. Our data suggest that zygotic genome activation occurs in two waves in Parhyale with microRNAs transcribed almost exclusively in the second wave. Contrary to findings in other arthropods, we do not predict a general role for microRNAs in clearing maternal transcripts. These data significantly expand the available transcriptomics resources for Parhyale, and facilitate its use as a model organism for the study of small RNAs in processes ranging from embryonic development to regeneration.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
High through-put sequencing of the Parhyale hawaiensis mRNAs and microRNAs to aid comparative developmental studies.PLoS One. 2012;7(3):e33784. doi: 10.1371/journal.pone.0033784. Epub 2012 Mar 20. PLoS One. 2012. PMID: 22448274 Free PMC article.
-
The maternal transcriptome of the crustacean Parhyale hawaiensis is inherited asymmetrically to invariant cell lineages of the ectoderm and mesoderm.PLoS One. 2013;8(2):e56049. doi: 10.1371/journal.pone.0056049. Epub 2013 Feb 13. PLoS One. 2013. PMID: 23418507 Free PMC article.
-
The amphipod crustacean Parhyale hawaiensis: An emerging comparative model of arthropod development, evolution, and regeneration.Wiley Interdiscip Rev Dev Biol. 2019 Sep;8(5):e355. doi: 10.1002/wdev.355. Epub 2019 Jun 11. Wiley Interdiscip Rev Dev Biol. 2019. PMID: 31183976 Free PMC article. Review.
-
Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis.Dev Biol. 2016 Jan 1;409(1):297-309. doi: 10.1016/j.ydbio.2015.10.029. Epub 2015 Nov 10. Dev Biol. 2016. PMID: 26569556
-
The maternal-zygotic transition: death and birth of RNAs.Science. 2007 Apr 20;316(5823):406-7. doi: 10.1126/science.1140693. Science. 2007. PMID: 17446392 Review.
Cited by
-
The transcriptional landscape of the giant freshwater prawn: Embryonic development and early sexual differentiation mechanisms.Front Endocrinol (Lausanne). 2022 Dec 7;13:1059936. doi: 10.3389/fendo.2022.1059936. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36568080 Free PMC article.
-
The maternal-to-zygotic transition: reprogramming of the cytoplasm and nucleus.Nat Rev Genet. 2025 Apr;26(4):245-267. doi: 10.1038/s41576-024-00792-0. Epub 2024 Nov 25. Nat Rev Genet. 2025. PMID: 39587307 Free PMC article. Review.
-
Molecular Approaches Detect Early Signals of Programmed Cell Death in Hippolyte inermis Leach.Curr Issues Mol Biol. 2024 Jun 18;46(6):6169-6185. doi: 10.3390/cimb46060368. Curr Issues Mol Biol. 2024. PMID: 38921039 Free PMC article.
References
-
- Martin A, et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Curr. Biol. 2016;26:14–26. - PubMed
-
- Browne WE, Price AL, Gerberding M, Patel NH. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 2005;42:124–149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
