

MIMOSA2: a metabolic network-based tool for inferring mechanism-supported relationships in microbiome-metabolome data
- PMID: 34999748
- PMCID: PMC8896604
- DOI: 10.1093/bioinformatics/btac003
MIMOSA2: a metabolic network-based tool for inferring mechanism-supported relationships in microbiome-metabolome data
Abstract
Motivation: Recent technological developments have facilitated an expansion of microbiome-metabolome studies, in which samples are assayed using both genomic and metabolomic technologies to characterize the abundances of microbial taxa and metabolites. A common goal of these studies is to identify microbial species or genes that contribute to differences in metabolite levels across samples. Previous work indicated that integrating these datasets with reference knowledge on microbial metabolic capacities may enable more precise and confident inference of microbe-metabolite links.
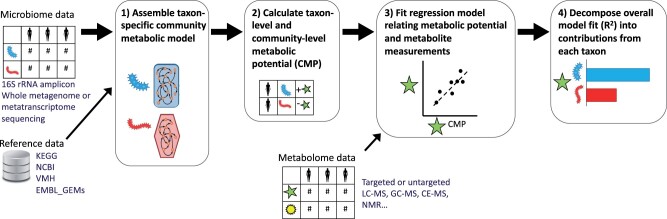
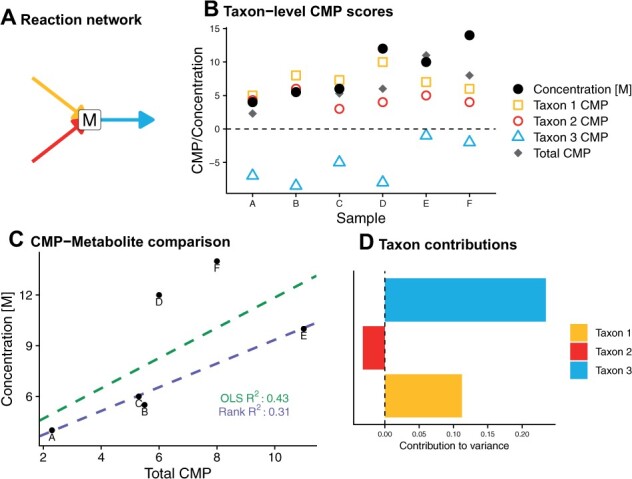
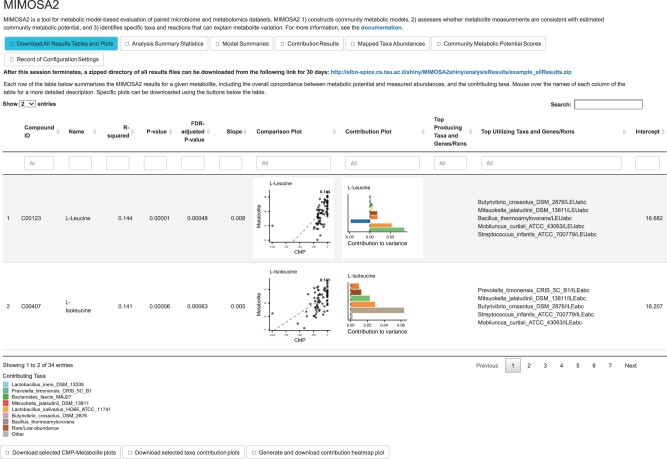
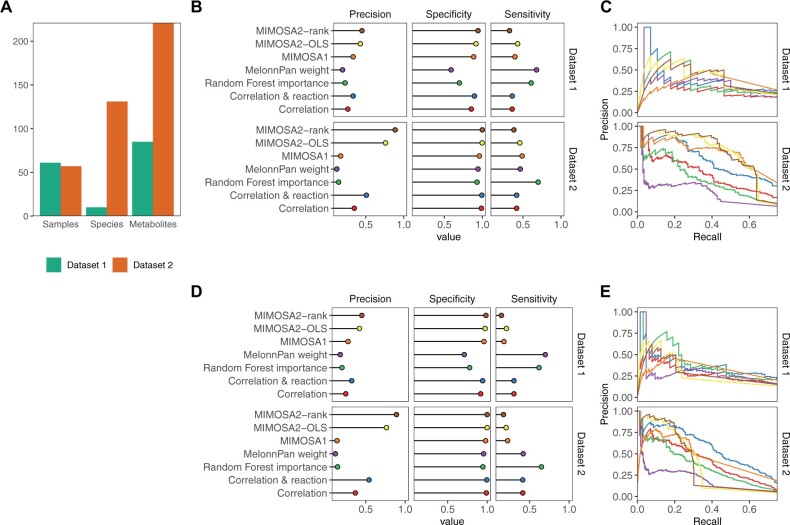
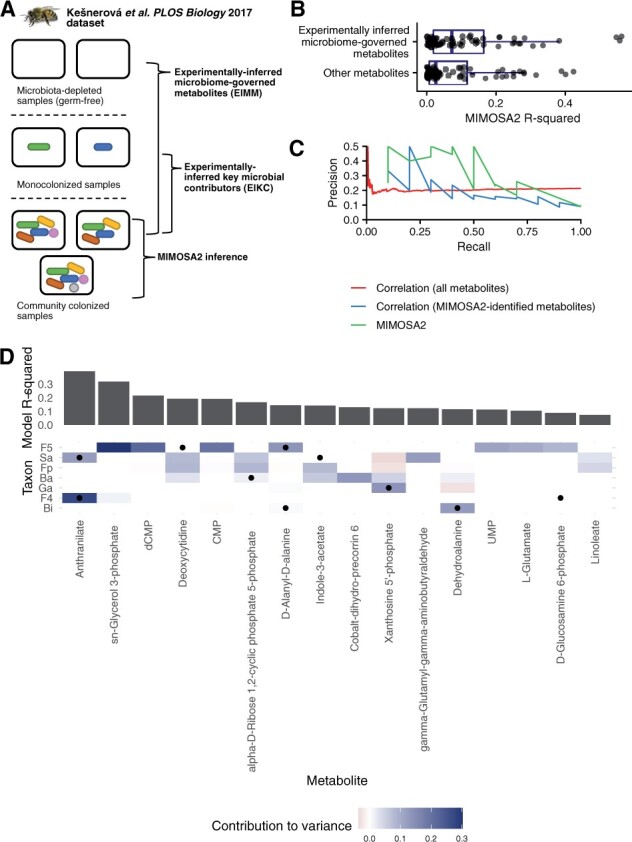
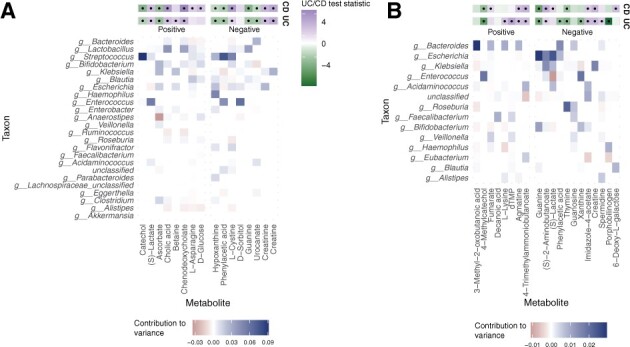
Results: We present MIMOSA2, an R package and web application for model-based integrative analysis of microbiome-metabolome datasets. MIMOSA2 uses genomic and metabolic reference databases to construct a community metabolic model based on microbiome data and uses this model to predict differences in metabolite levels across samples. These predictions are compared with metabolomics data to identify putative microbiome-governed metabolites and taxonomic contributors to metabolite variation. MIMOSA2 supports various input data types and customization with user-defined metabolic pathways. We establish MIMOSA2's ability to identify ground truth microbial mechanisms in simulation datasets, compare its results with experimentally inferred mechanisms in honeybee microbiota, and demonstrate its application in two human studies of inflammatory bowel disease. Overall, MIMOSA2 combines reference databases, a validated statistical framework, and a user-friendly interface to facilitate modeling and evaluating relationships between members of the microbiota and their metabolic products.
Availability and implementation: MIMOSA2 is implemented in R under the GNU General Public License v3.0 and is freely available as a web server at http://elbo-spice.cs.tau.ac.il/shiny/MIMOSA2shiny/ and as an R package from http://www.borensteinlab.com/software_MIMOSA2.html.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Adamovsky O. et al. (2020) Evaluation of microbiome-host relationships in the zebrafish gastrointestinal system reveals adaptive immunity is a target of bis(2-ethylhexyl) phthalate (DEHP) exposure. Environ. Sci. Technol., 54, 5719–5728. - PubMed
-
- Bourgonje A.R. et al. (2020) Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends Mol. Med., 26, 1034–1046. - PubMed
